-
DARTE-QM
Effective monitoring of antibiotic resistance genes and their dissemination in environmental ecosystems has been hindered by the cost and efficiency of methods available for the task. We developed a method entitled the Diversity of Antibiotic Resistance genes and Transfer Elements-Quantitative Monitoring (DARTE-QM), a system implementing high-throughput sequencing to simultaneously sequence thousands of antibiotic resistant genes representing a full-spectrum of antibiotic resistance classes commonly seen inenvironmental systems. In this study, we demonstrated DARTE-QM by screening 367 antibiotic resistance genes within environmental samples originated from manure, soil, and animal feces, in addition to a mock-community used as a control to test performance.DARTE-QM offers a supplemental approach to studying antibiotic resistance in environmental microbiomes, showing advantages in efficiency and the ability to scale for many samples. This method provides a means of data acquisitionthat will alleviate the obstacles that many researchers in this area face.
-
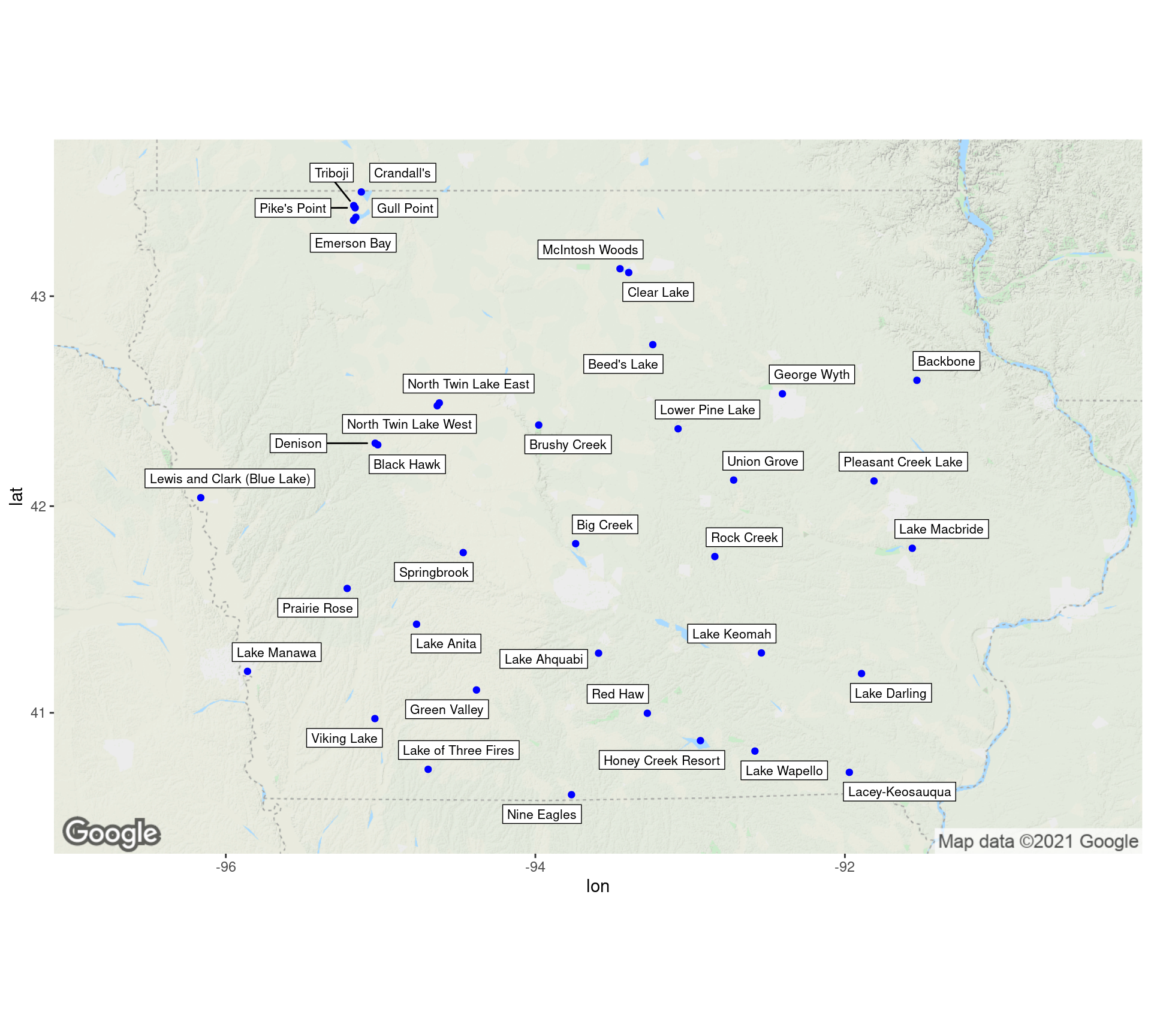
Harmful Algal Blooms in Iowa Lakes
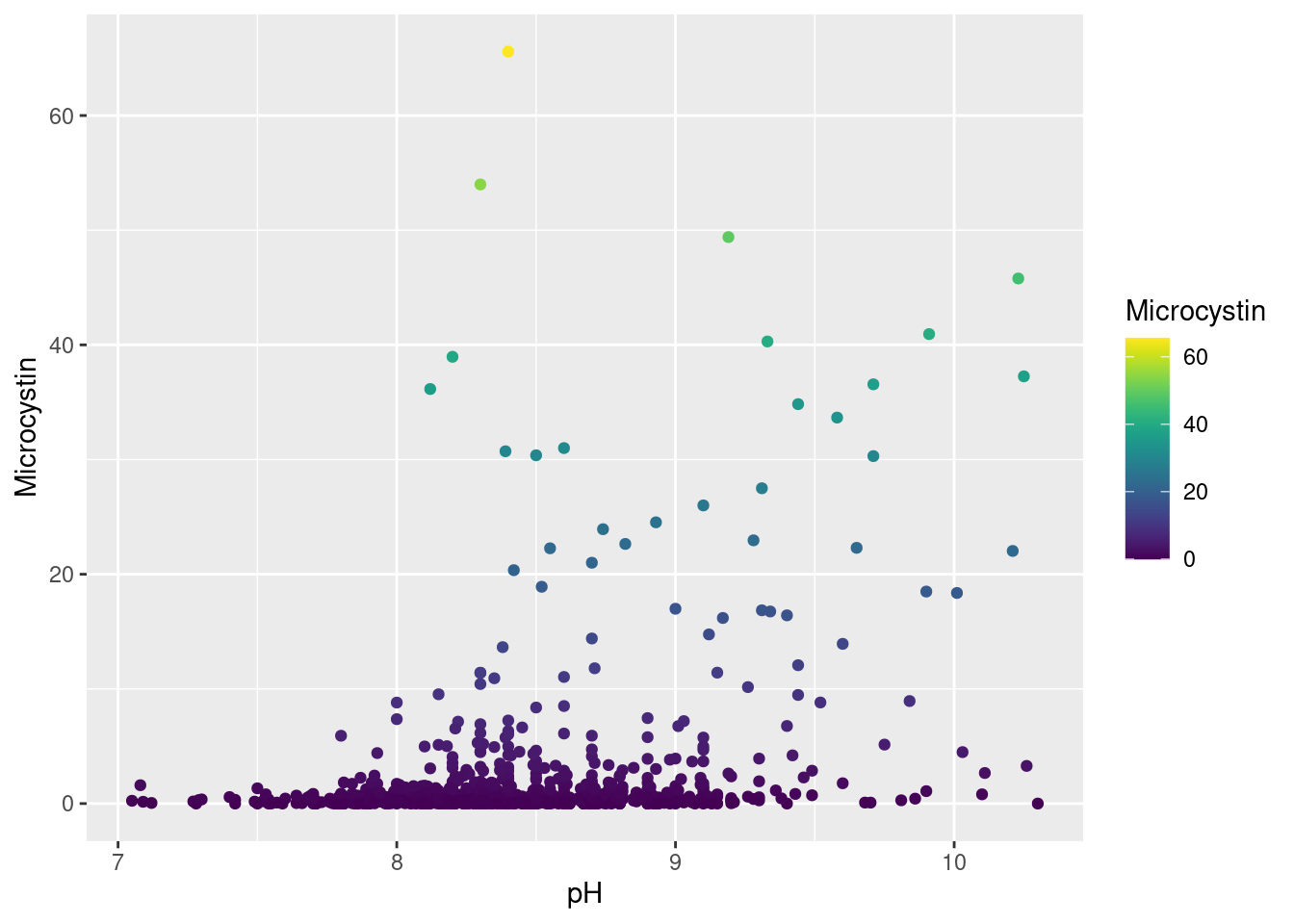
-
phylosmith: an R-package for reproducible and efficient microbiome analysis
phylosmith is a conglomeration of functions written to process and analyze phyloseq-class objects. Phyloseq objects are a great data-standard for microbiome, gene-expression, and many other data types.
A lot of these functions are just to make “data-wrangling” easier for the user. Others will implement complex routines in a, hopefully, efficient and concise manner. I have also made functions to make figures for quick examination of data, but they may or may not be suitable for publication as some may require parameter optimization.
-
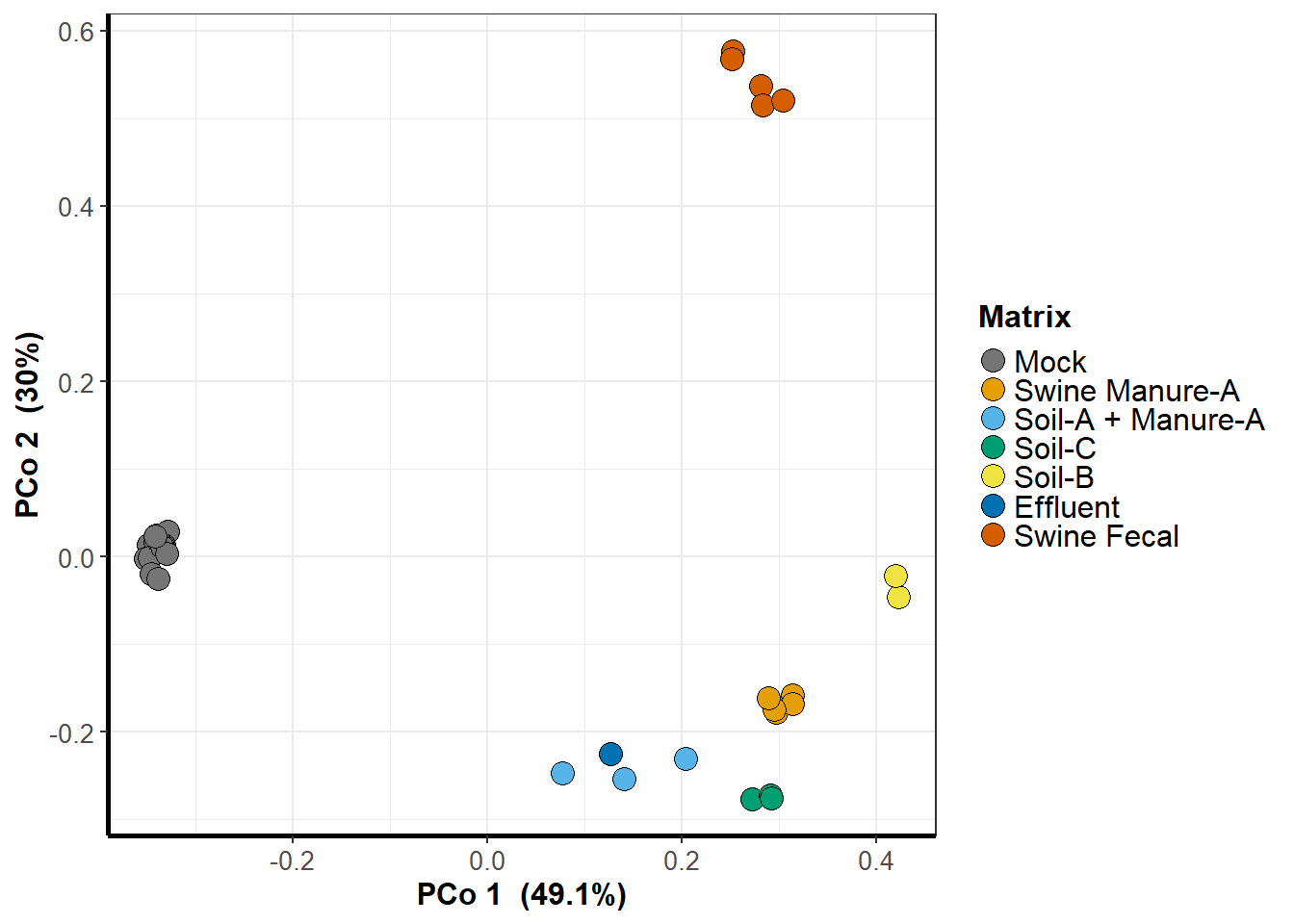
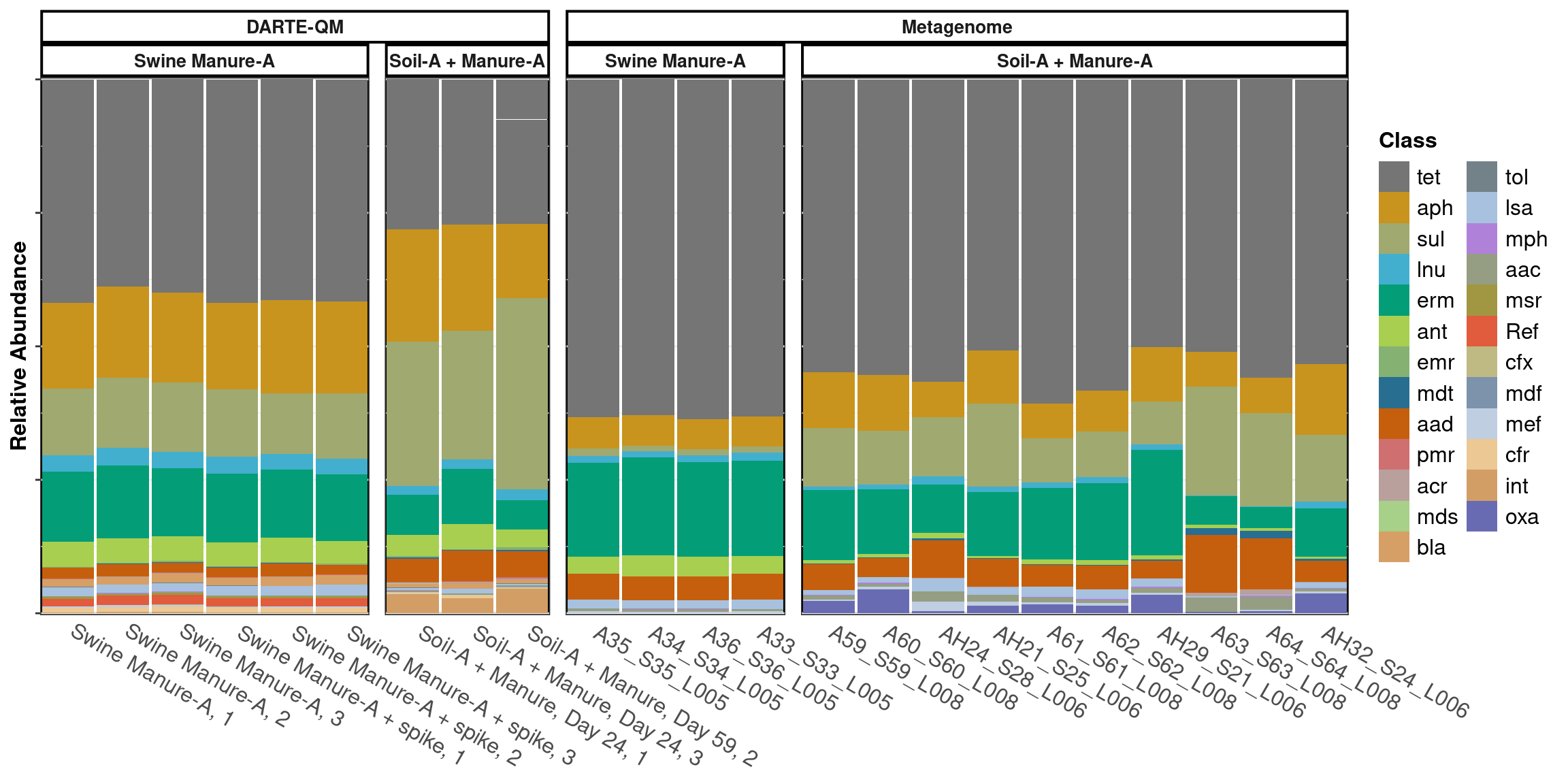
Investigating the dispersal of antibiotic resistance associated genes from manure application to soil and drainage waters in simulated agricultural farmland systems
Manure from animals that have been treated with antibiotics is often used to fertilize agricultural soils and its application has previously been shown to enrich for genes associated with antibiotic resistance in agroecosystems. To investigate the magnitude of this effect, we designed a column experiment simulating manure-treated agricultural soil that utilizes artificial subsurface drainage to determine the duration and extent which this type of manure fertilization impacts the set of genes associated with antibiotic resistance in drainage water. We classified ARGs in manure-treated drainage effluent water by its source of origin. Overall, we found that 61% and 7% of the total abundance of ARGs found in drainage water samples could be attributed to manure enrichment and manure addition, respectively. Among these ARGs, we identified 75 genes unique to manure that persisted in both soil and drainage water throughout a drainage season typical of the Upper Midwestern United States. While most of these genes gradually decreased in abundance over time, the IS6100-associated tet(33) gene accrued. These results demonstrate the influence of manure applications on the composition of the resistome observed in agricultural drainage water and highlight the importance of anthropogenic ARGs in the environment.


-
Development and application of a bioinformatics pipeline for genotyping-by-sequencing (GBS) of autotetraploid potato
Genotyping-by-sequencing (GBS) is being widely used in diploid crops as an efficient technology for identifying genome-wide markers to assist breeding. Both genome-wide association studies and genomic selection have benefited from the availability of GBS markers (Morris et al. 2012; Poland et al. 2012). One of the challenges with GBS is how to handle missing data, but for diploids there are many accurate and accessible options for marker imputation (Rutkoski et al. 2013; Swarts et al. 2014). For autotetraploid crops, such as potato (Solanum tuberosum), the cost effectiveness of GBS is less certain due to the higher read depths (50–60X) needed to accurately differentiate the three heterozygous genotypes. There is also less information published about imputation methods and accuracy. Our objectives were to (1) develop a bioinformatics pipeline for GBS variant discovery, genotype calling, and imputation in autotetraploids, (2) apply the pipeline to a panel of elite potato breeding lines and varieties from across North America, and (3) interpret the results in the context of a competing marker technology: the potato Infinium SNP array (Douches et al. 2014).


-
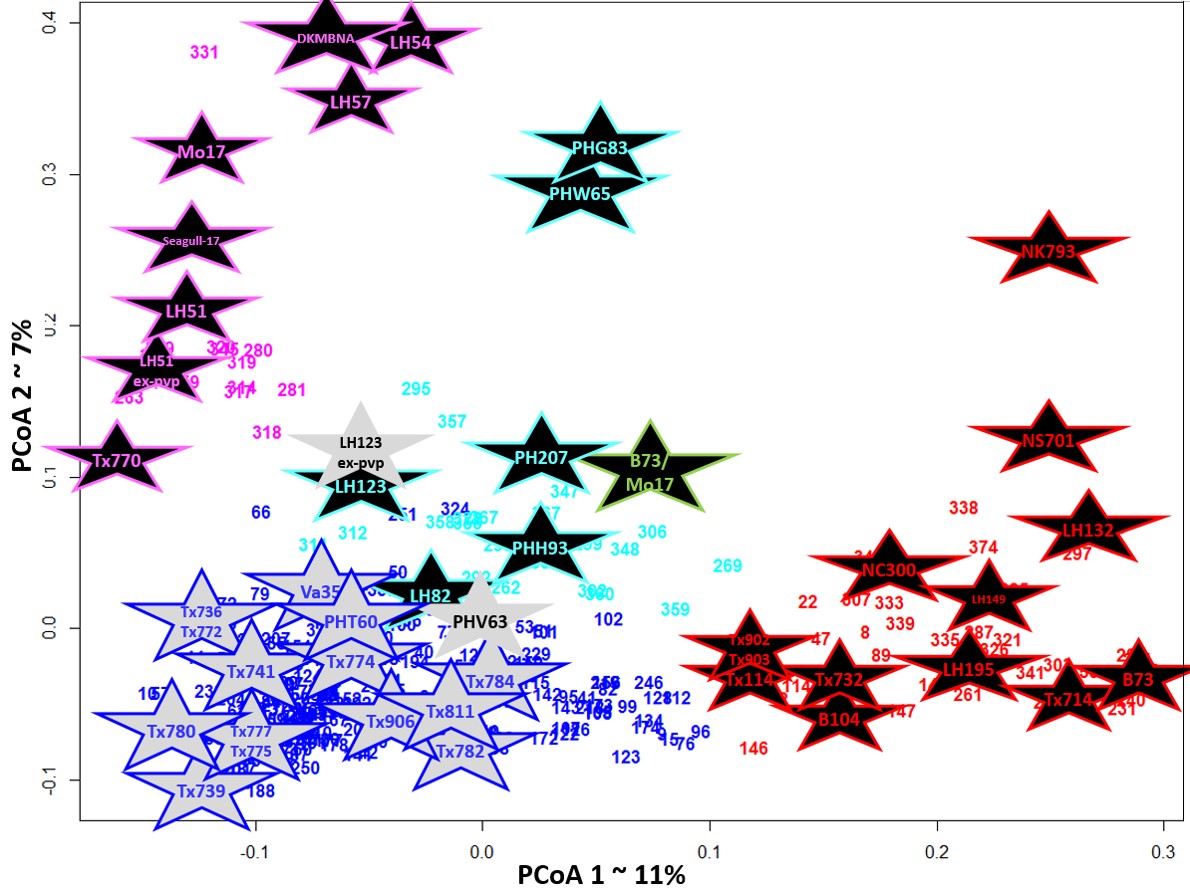
Molecular analysis of genetic diversity in a Texas maize (Zea maysL) breeding program
The Texas maize (Zea mays L) breeding program at Texas A&M University has been unique among breeding pro-grams for the incorporation of diverse germplasm from a wide range of origins into elite inbred lines. The Texas program, situated in a subtropical environment, has found beneficial traits in maize of tropical origin beyond what is available in the temperate material commonly used in the far more productive Midwestern region of the United States. To date, no molecular studies had been conducted to make any quantitative differentiations between the genetic diversity in the germplasm developed in the Texas program or comparisons to the germplasm available from the Midwest. In this study, a molecular characterization of genetic diversity was performed. A unique set of 266 elite Texas lines were genotyped using 766 single nucleotide polymorphism markers, this was then combined with data published in a previous study focusing on ex-PVP lines released by private companies. The two data sets combined had 380 genotypes with 635 markers. It was determined that there were five subpopulations of material in this combined set as demonstrated by population structure. The data suggested that the array mark-ers, designed to cluster the Midwestern heterotic groups, did not discriminate this exotic material well and/or that the Texas heterotic pools were not well supported. We conclude that the majority of Texas program material is a novel population, genetically dissimilar to Midwest temperate material, and would be a useful source of unique genetics for other maize breeding programs