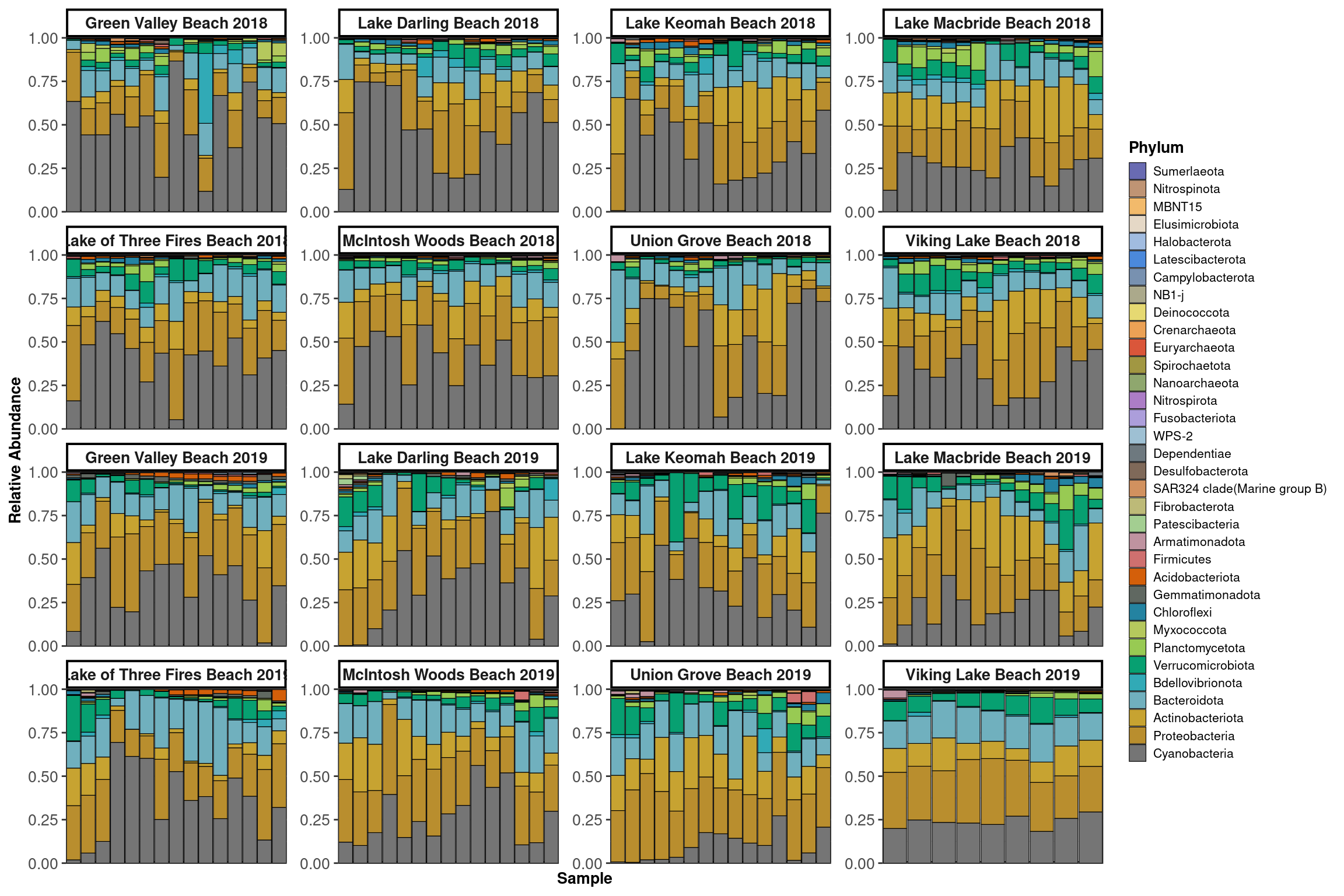
Communities
Profiles
for (risk in c('Low', 'High')){
cat('###', risk, '<br>', '\n\n')
profile <- phylosmith::phylogeny_profile(taxa_filter(lake_hl, "Lake_Risk",
risk), classification = 'Phylum', treatment = c('Location', 'Year'),
grid = TRUE, relative_abundance = TRUE)
print(profile)
cat('\n', '<br><br><br>', '\n\n')
}Low
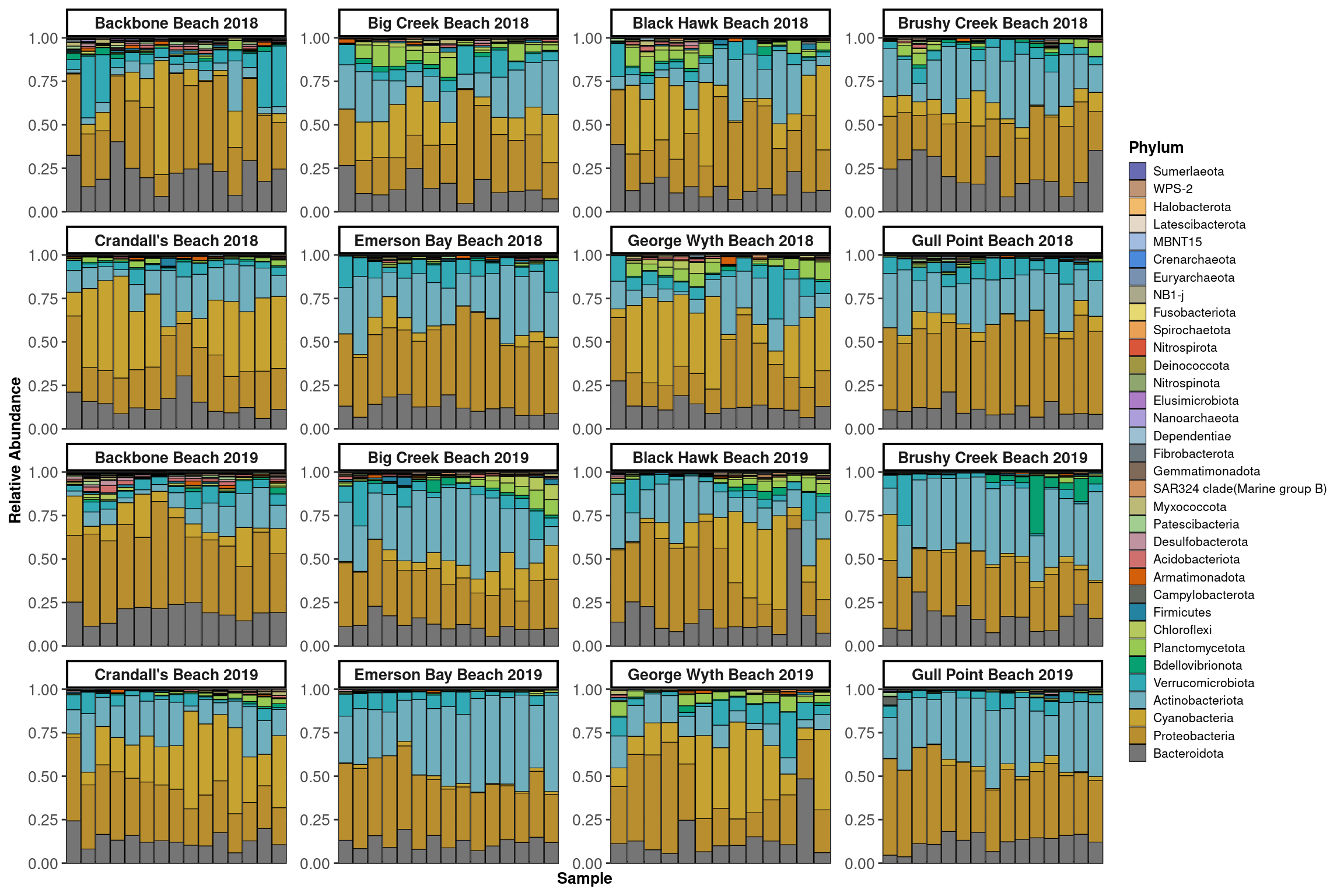
High
Co-Occurrences
Calculate
lake_hl_co <- taxa_filter(conglomerate_taxa(lake_hl, "Genus", TRUE,
use_taxonomic_names = FALSE), c("Location", "Year"), frequency = 0.25)
lake_hl_co <- co_occurrence(lake_hl_co, treatment = c("Location", "Year"))
lake_co_table <- lake_hl_co[p < 0.05]
setorder(lake_co_table, -rho)Seperate High-Low
lake_co_table <- lake_co_table[rho < 0]
lake_co_table[,p:=NULL]
lake_co_table[, Risk := "Low"]
set(lake_co_table, unlist(sapply(high_risk_lakes, FUN=function(x)grep(x,lake_co_table$Treatment))), "Risk", "High")
lake_co_table <- lake_co_table[, c("X", "Y", "Treatment", "Risk", "rho")]
lake_co_table[,Edge := paste(X,Y,sep="-")]
high <- lake_co_table[unlist(sapply(high_risk_lakes, FUN=function(x)grep(x,lake_co_table$Treatment))),]
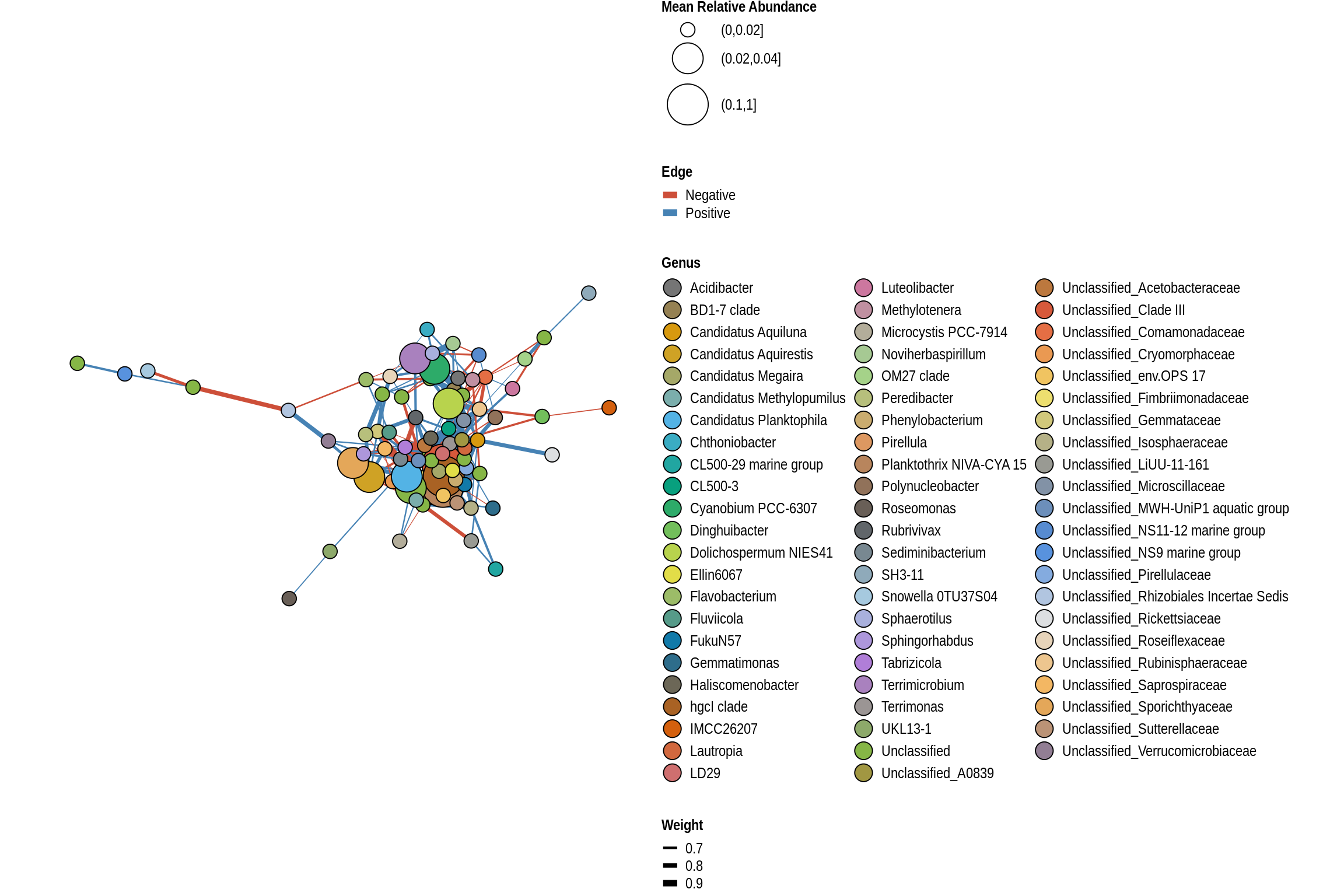
low <- lake_co_table[unlist(sapply(low_risk_lakes, FUN=function(x)grep(x,lake_co_table$Treatment))),]Lake Networks
phyloseq_obj <- relative_abundance(taxa_filter(conglomerate_taxa(lake_hl,
"Genus", TRUE, FALSE), c("Location", "Year"), frequency = 0.25))
for (year in unique(metadata$Year)){
cat('### ', year, '<br>', '\n\n')
cat('#### Lake {.tabset}', '\n', '<br>', '\n\n')
for (lake in unique(phyloseq_obj@sam_data$Location)){
cat('#####', lake, '<br>', '\n\n')
phylo_obj <- taxa_filter(phyloseq_obj, c("Location","Year"), c(lake), frequency = 0.5)
phylo_obj <- taxa_filter(phylo_obj, c("Year"), year, frequency = 0.7)
net <- co_occurrence_network(phylo_obj, classification = "Genus")
print(net + guides(fill = guide_legend(ncol = 3, override.aes = list(size = 5))))
cat('\n', '<br><br><br>', '\n\n')
}
cat('\n', '<br><br><br>', '\n\n')
}2018
Lake
George Wyth Beach
McIntosh Woods Beach

Backbone Beach
Gull Point Beach
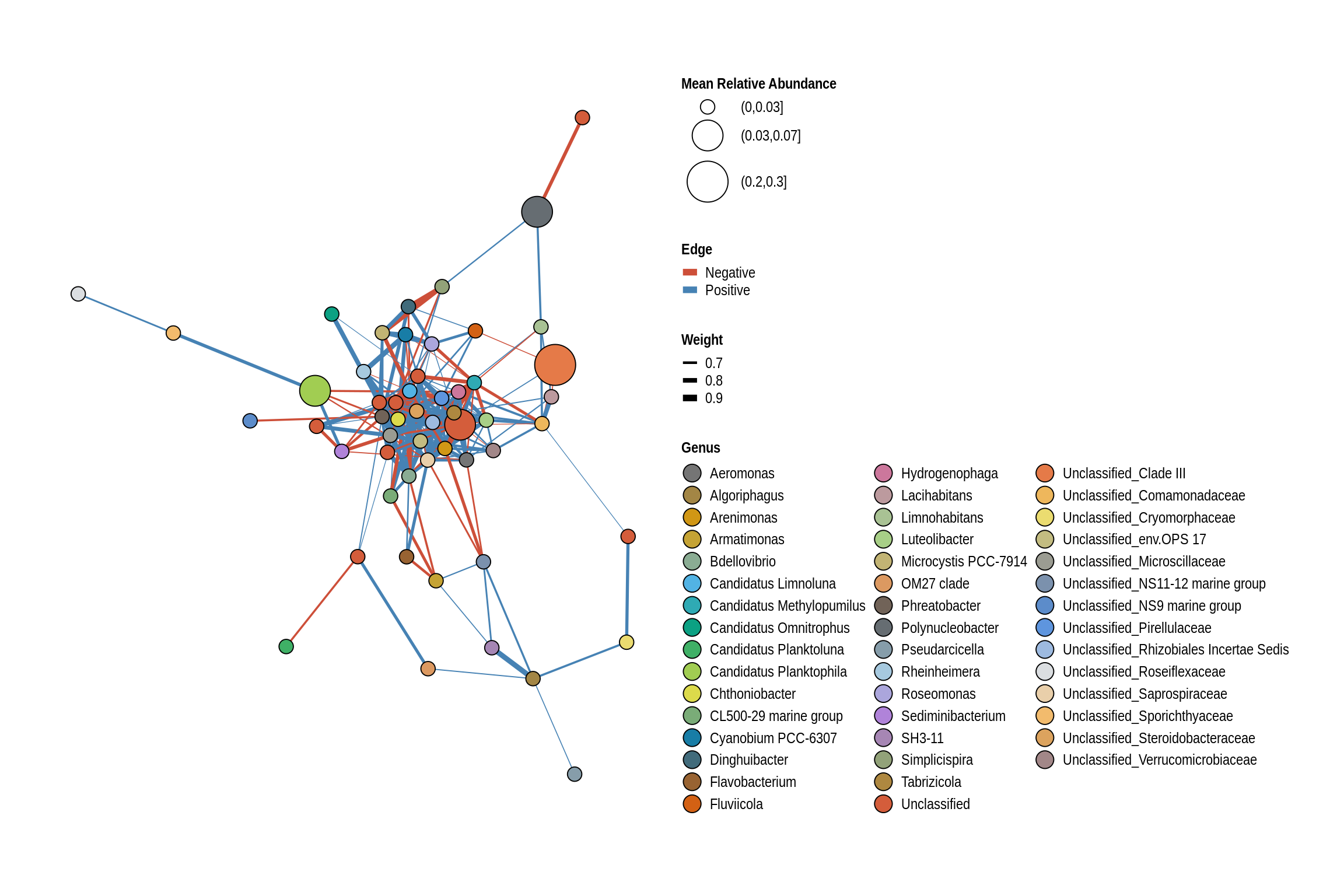
Emerson Bay Beach

Crandall’s Beach

Lake Macbride Beach
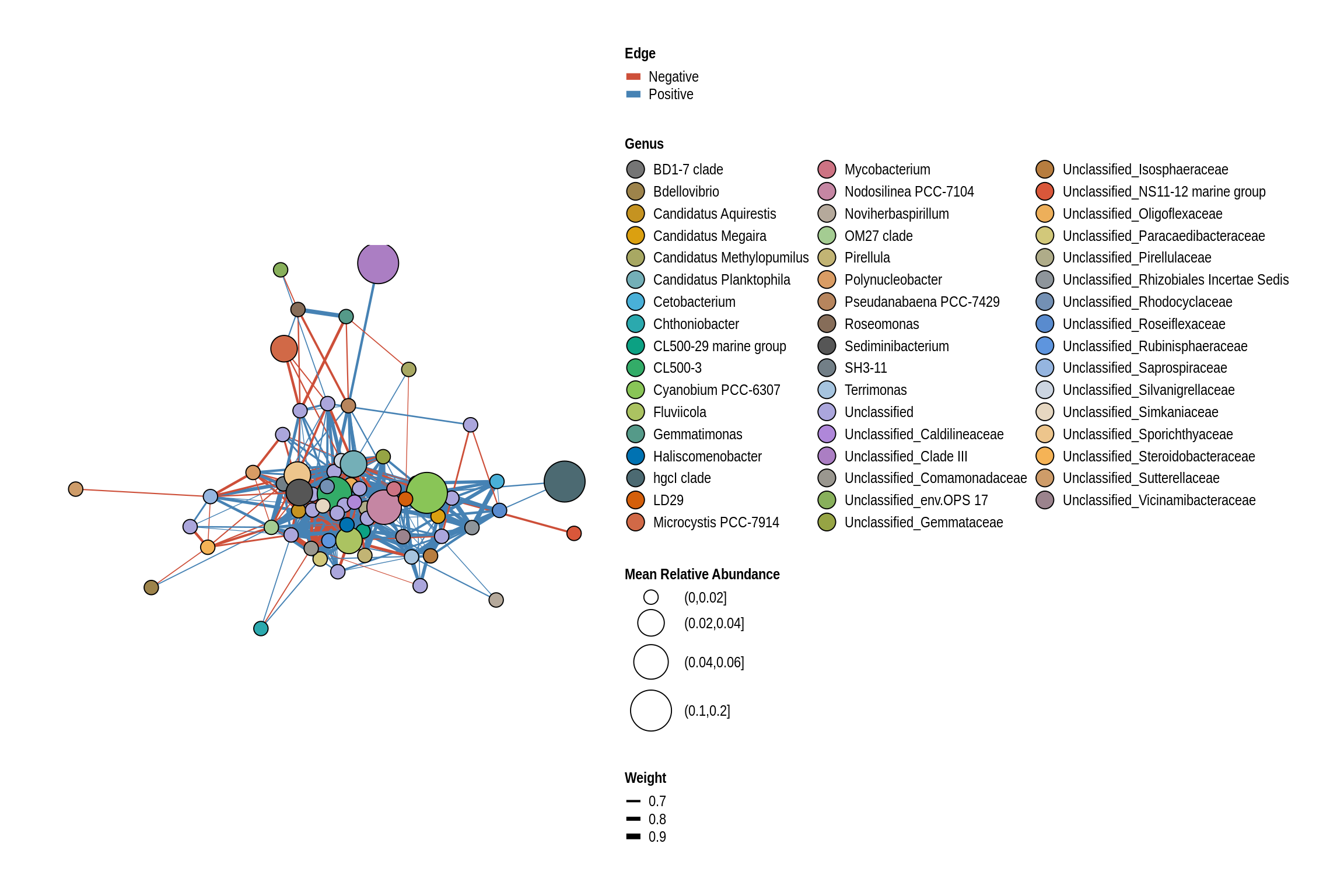
Lake Keomah Beach
Viking Lake Beach
Big Creek Beach

Black Hawk Beach

Union Grove Beach

Lake of Three Fires Beach

Green Valley Beach
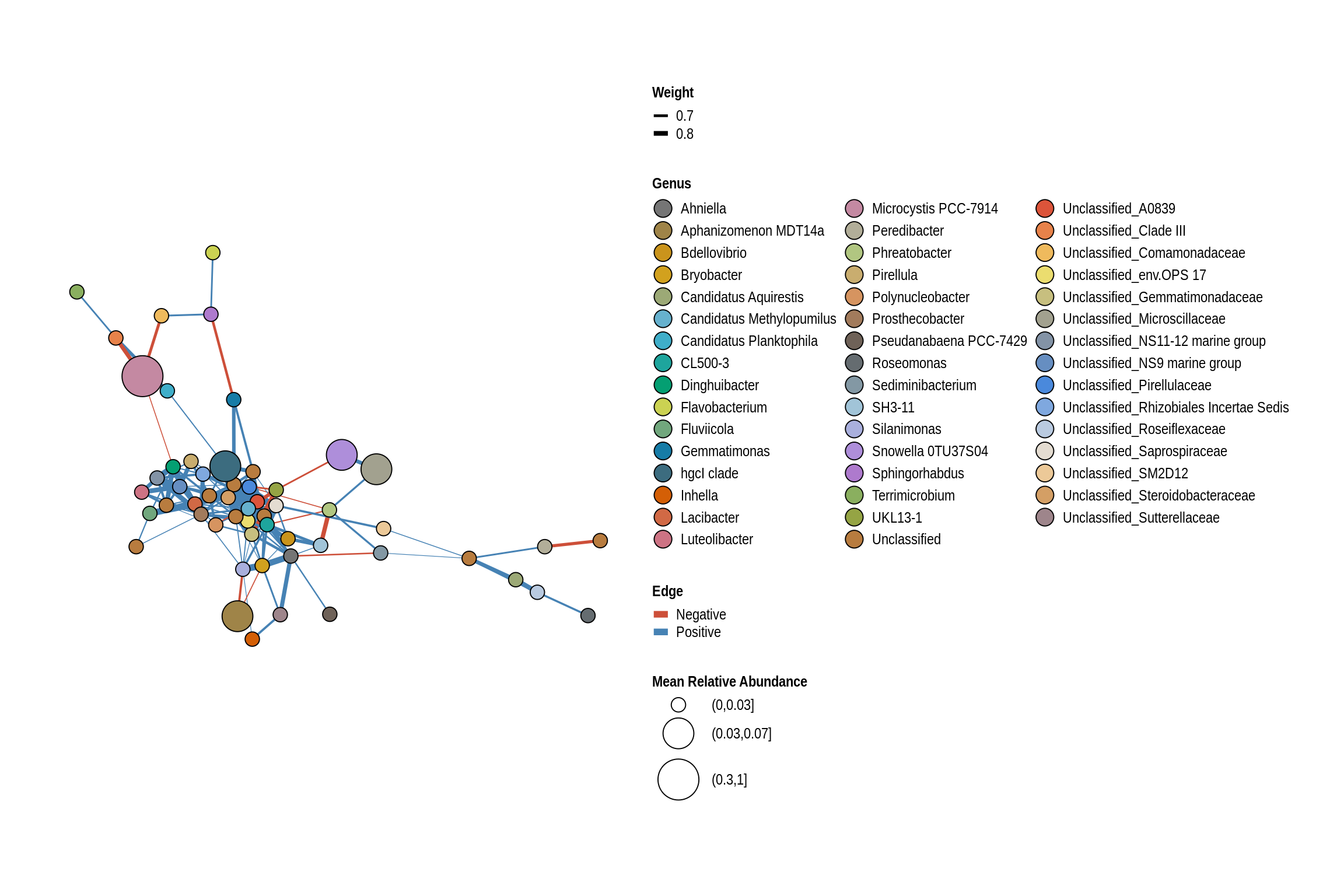
Lake Darling Beach
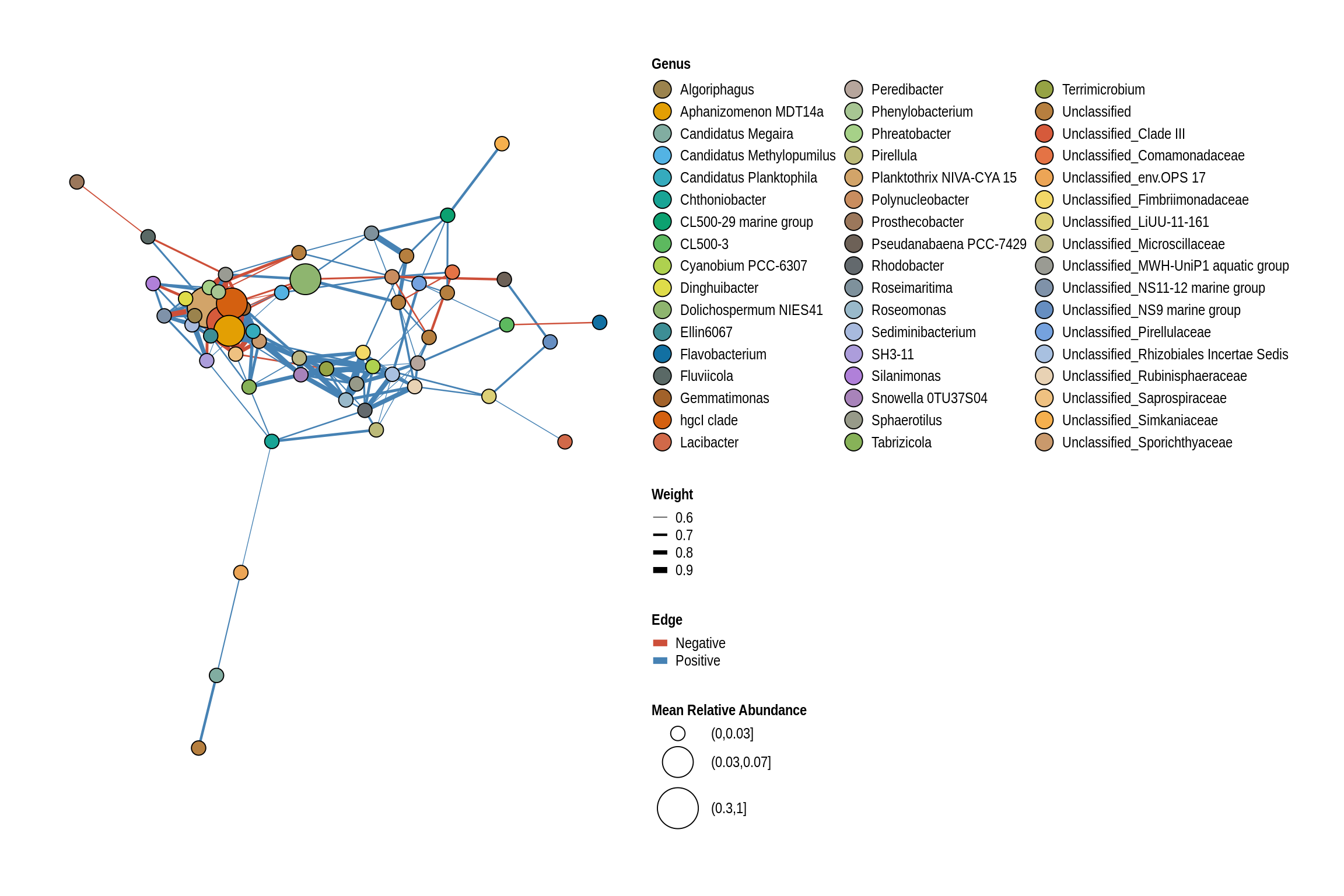
Brushy Creek Beach

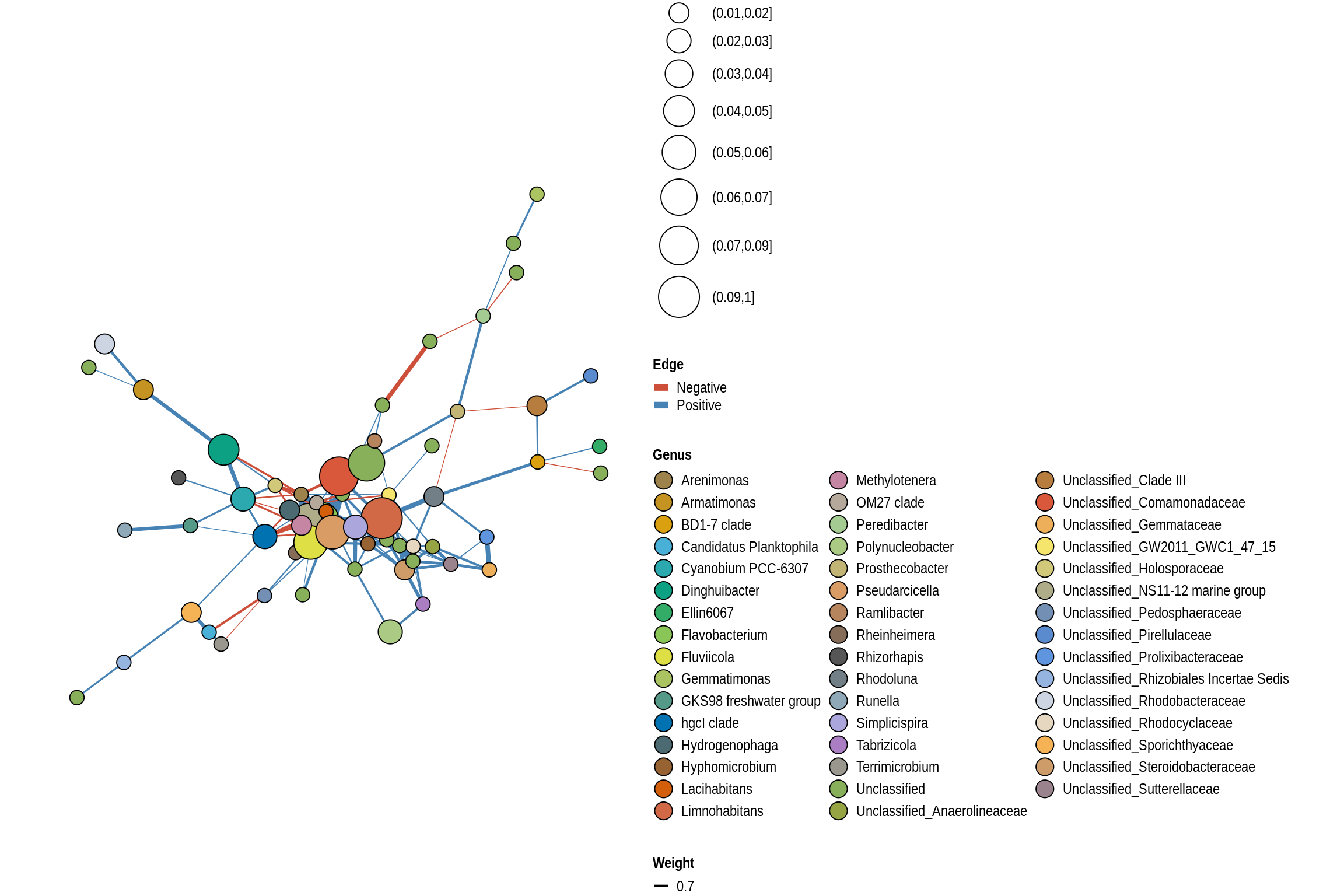
2019
Lake
George Wyth Beach
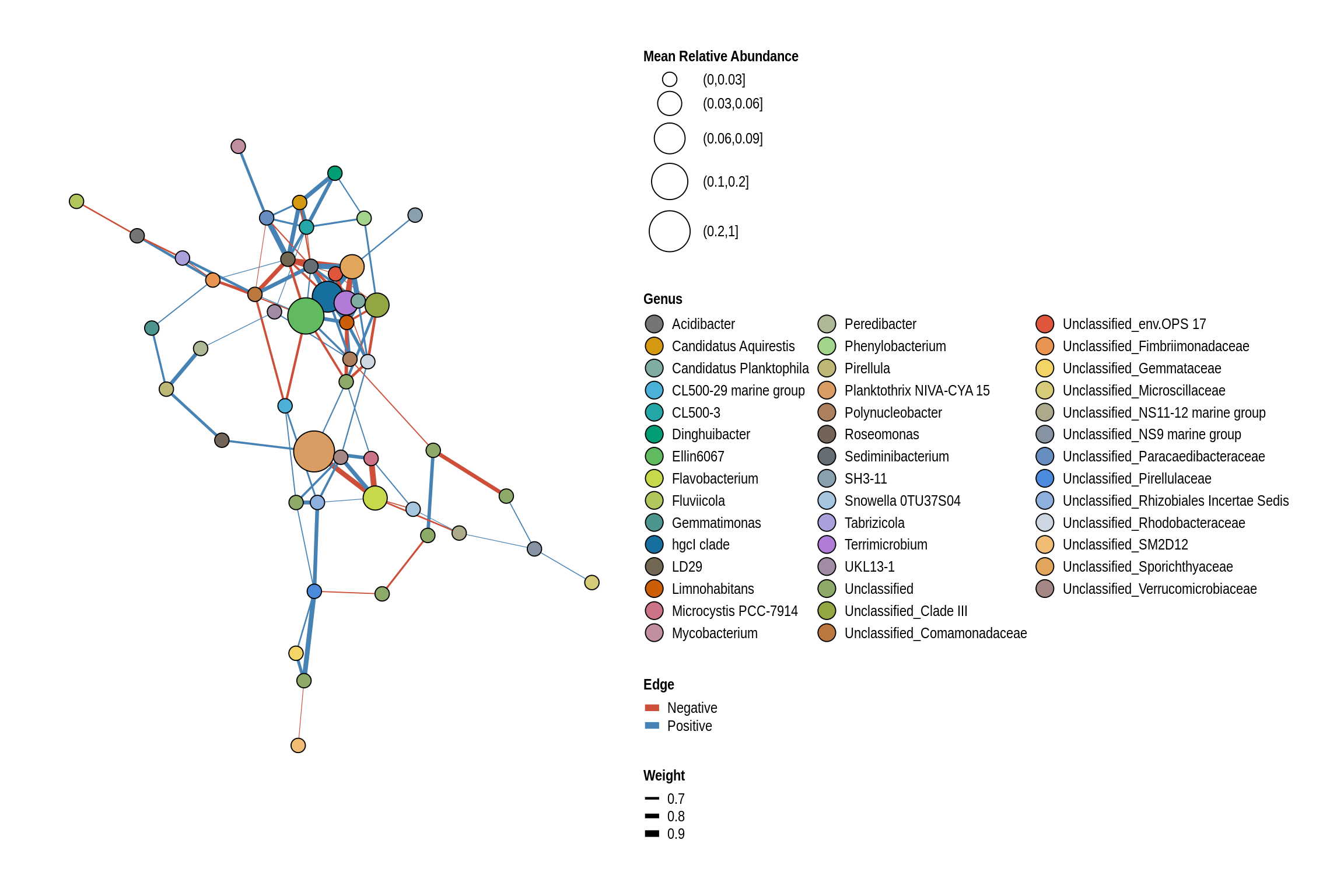
McIntosh Woods Beach
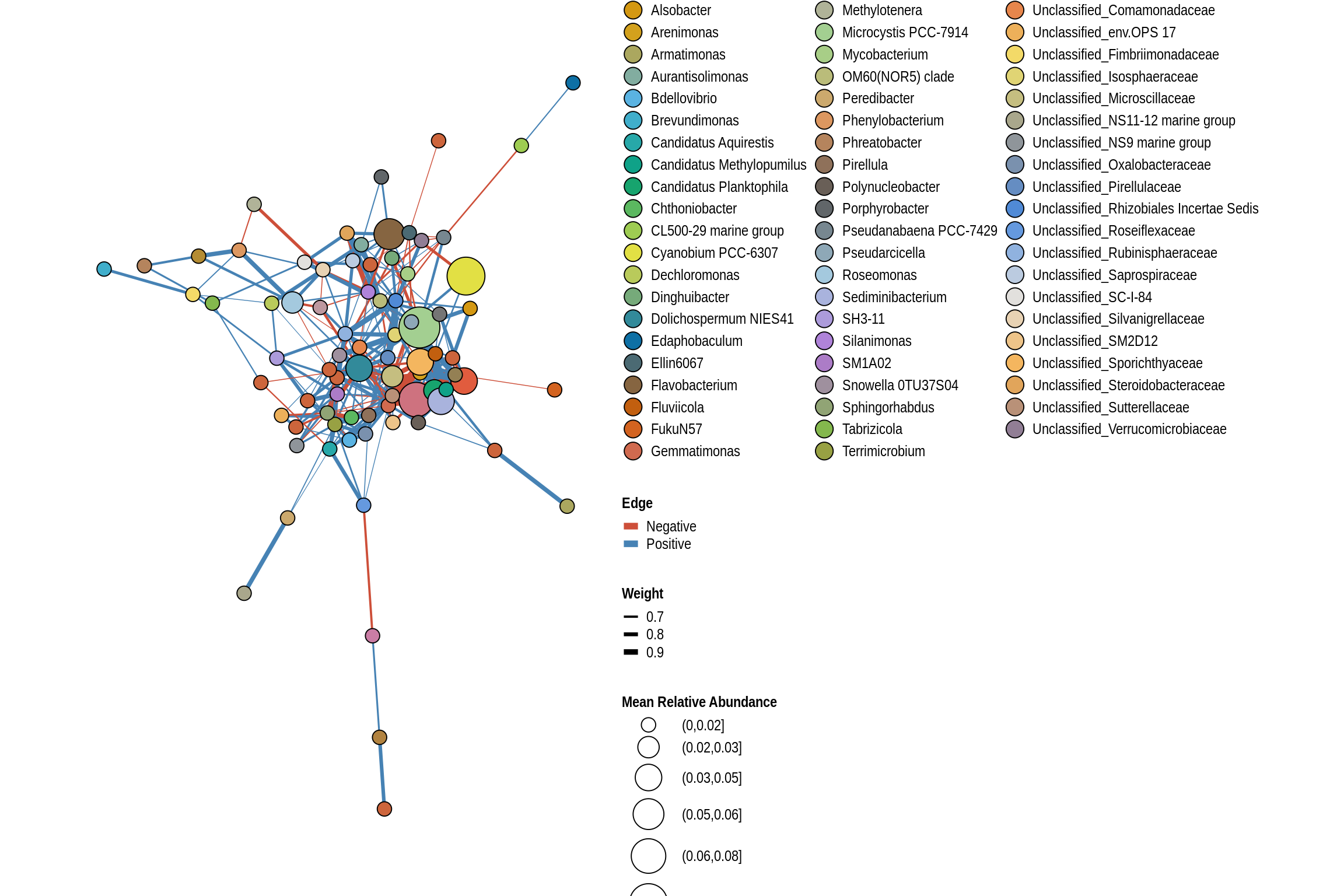
Backbone Beach

Gull Point Beach

Emerson Bay Beach
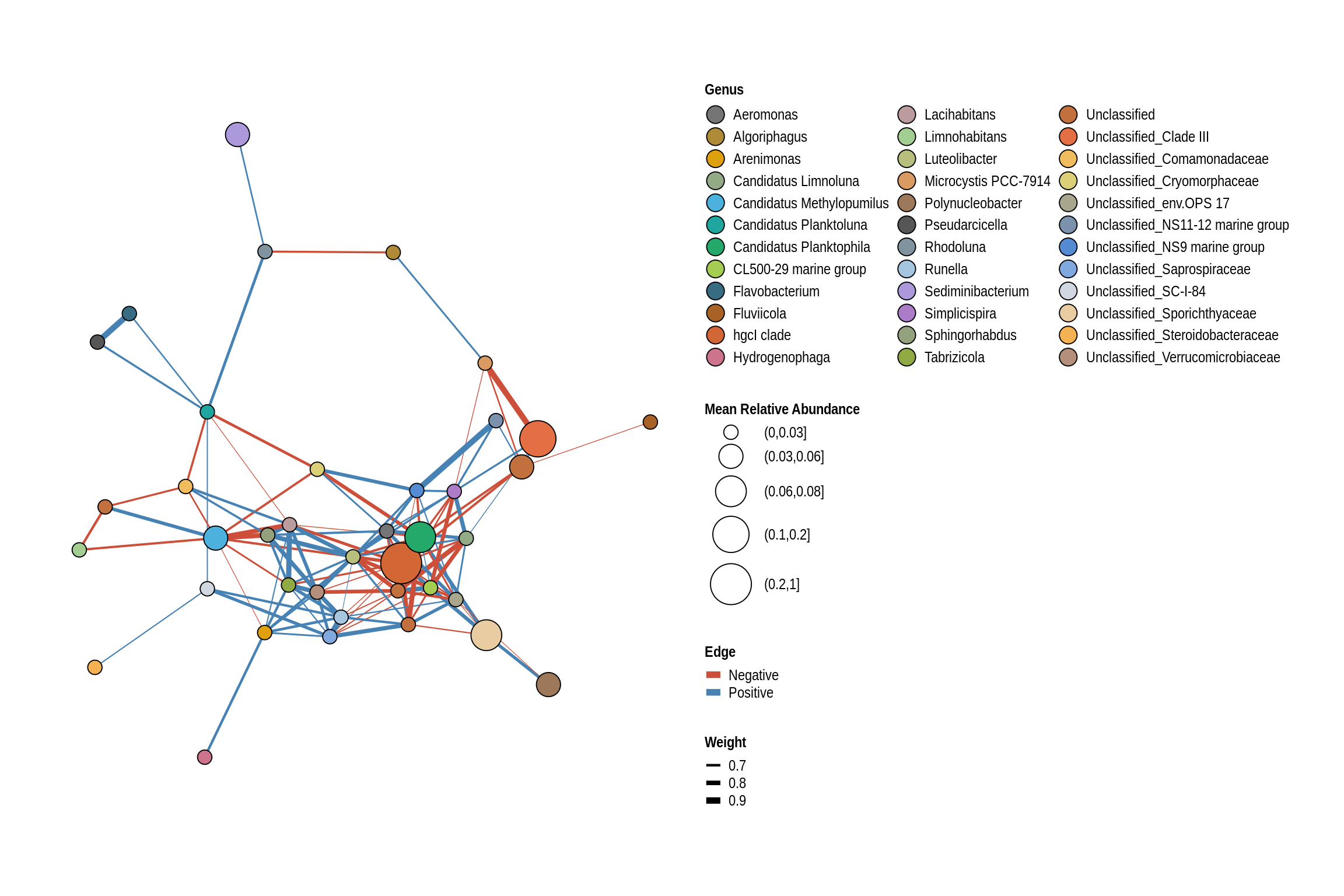
Crandall’s Beach
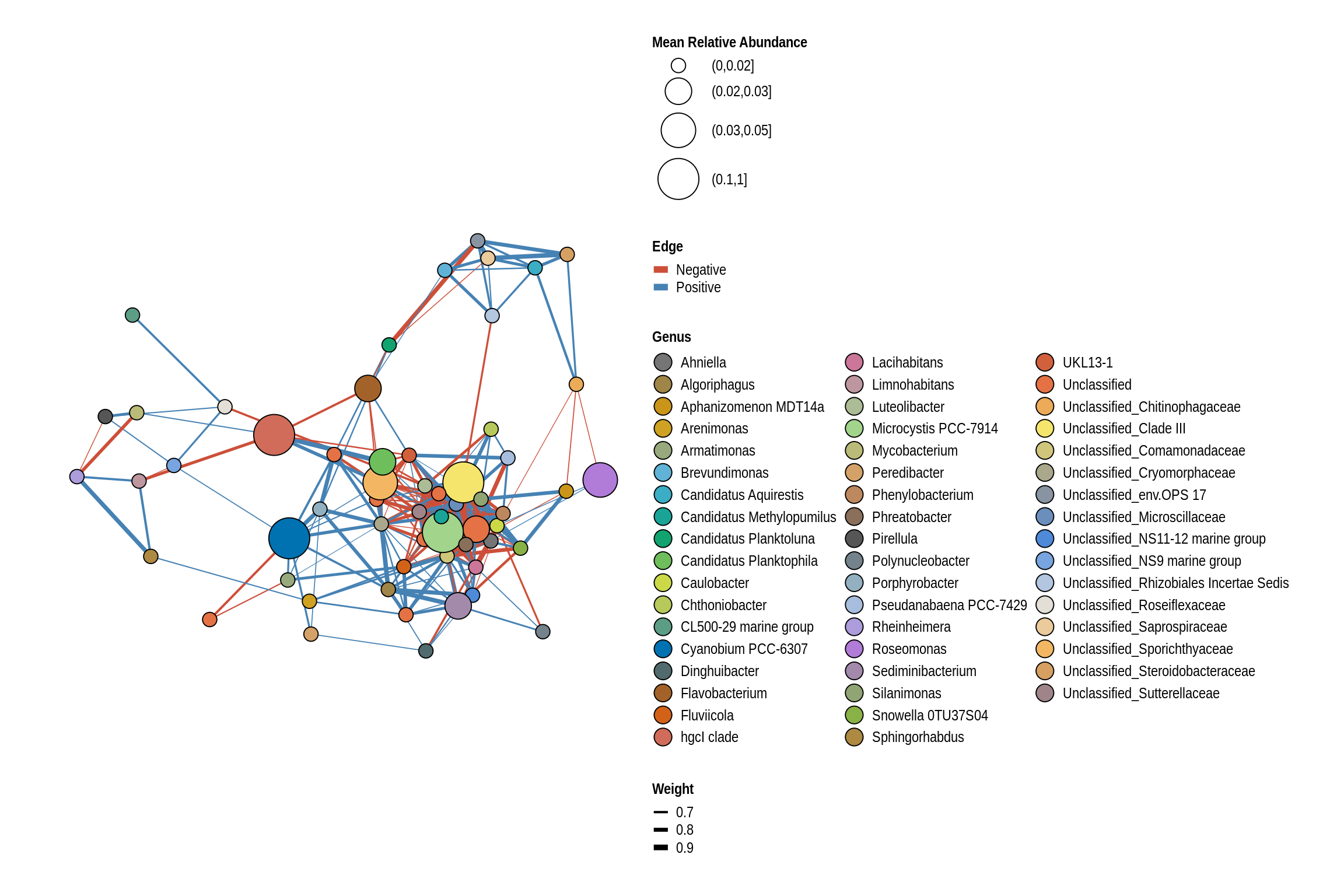
Lake Macbride Beach

Lake Keomah Beach
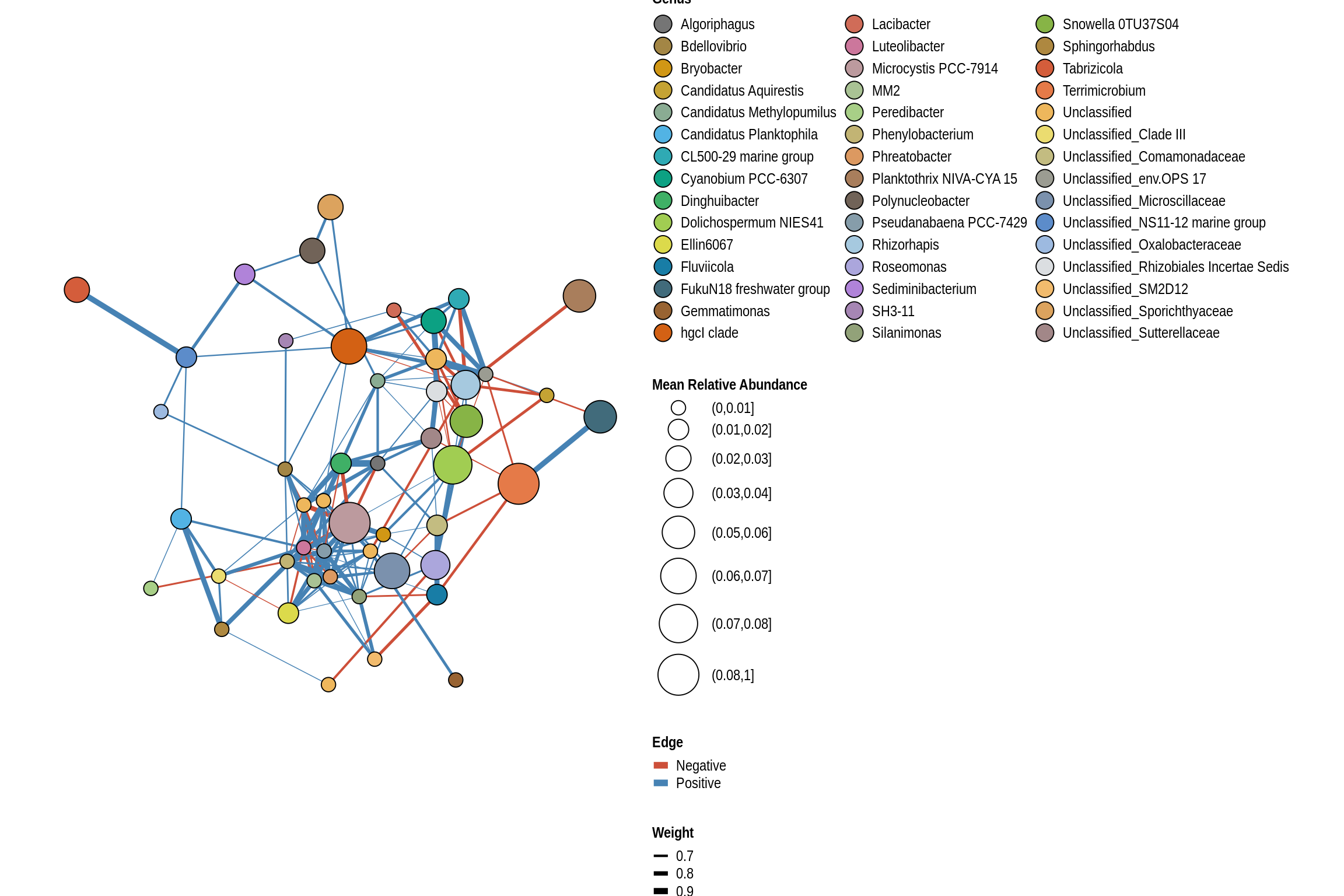
Viking Lake Beach

Big Creek Beach
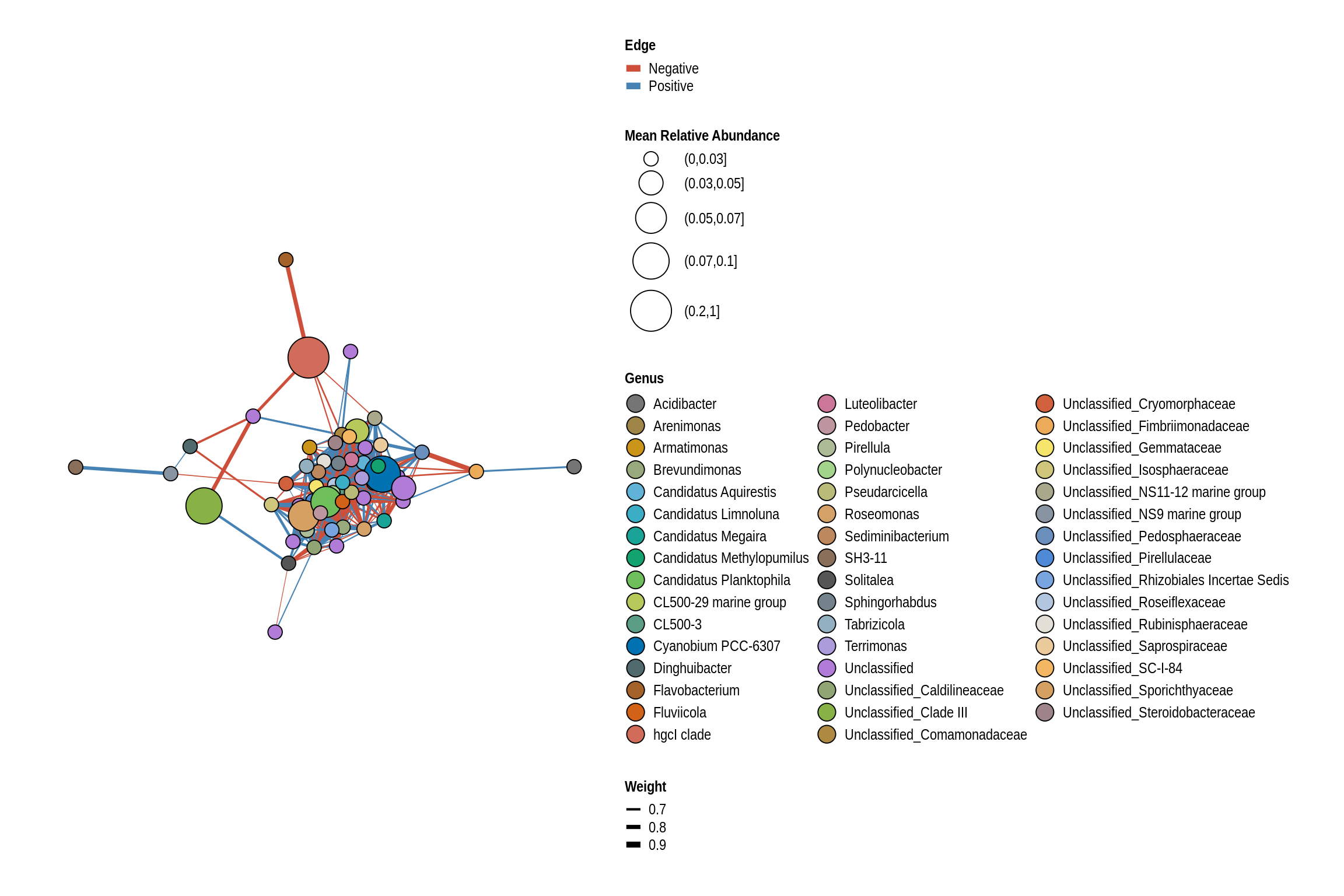
Black Hawk Beach
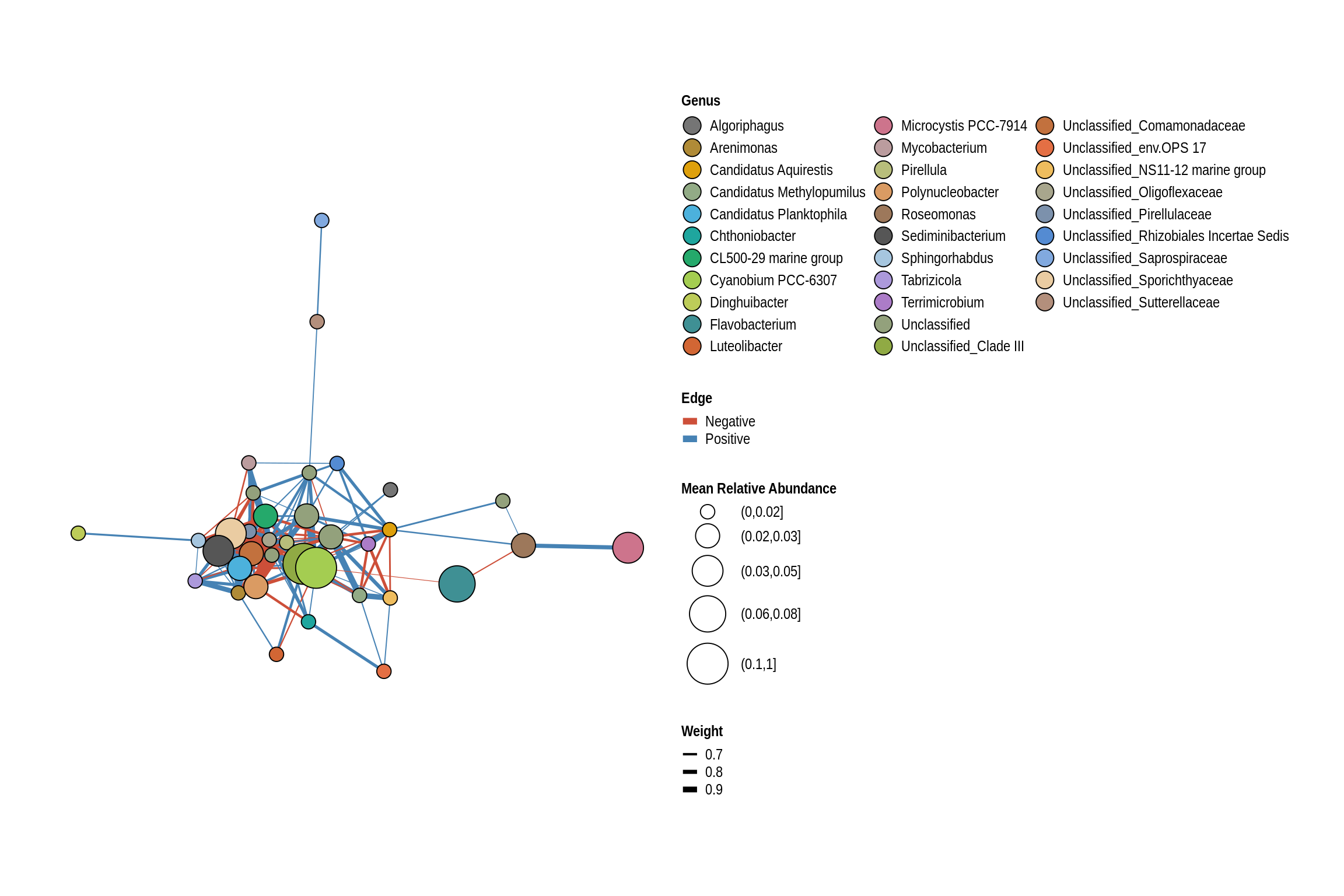
Union Grove Beach

Lake of Three Fires Beach

Green Valley Beach
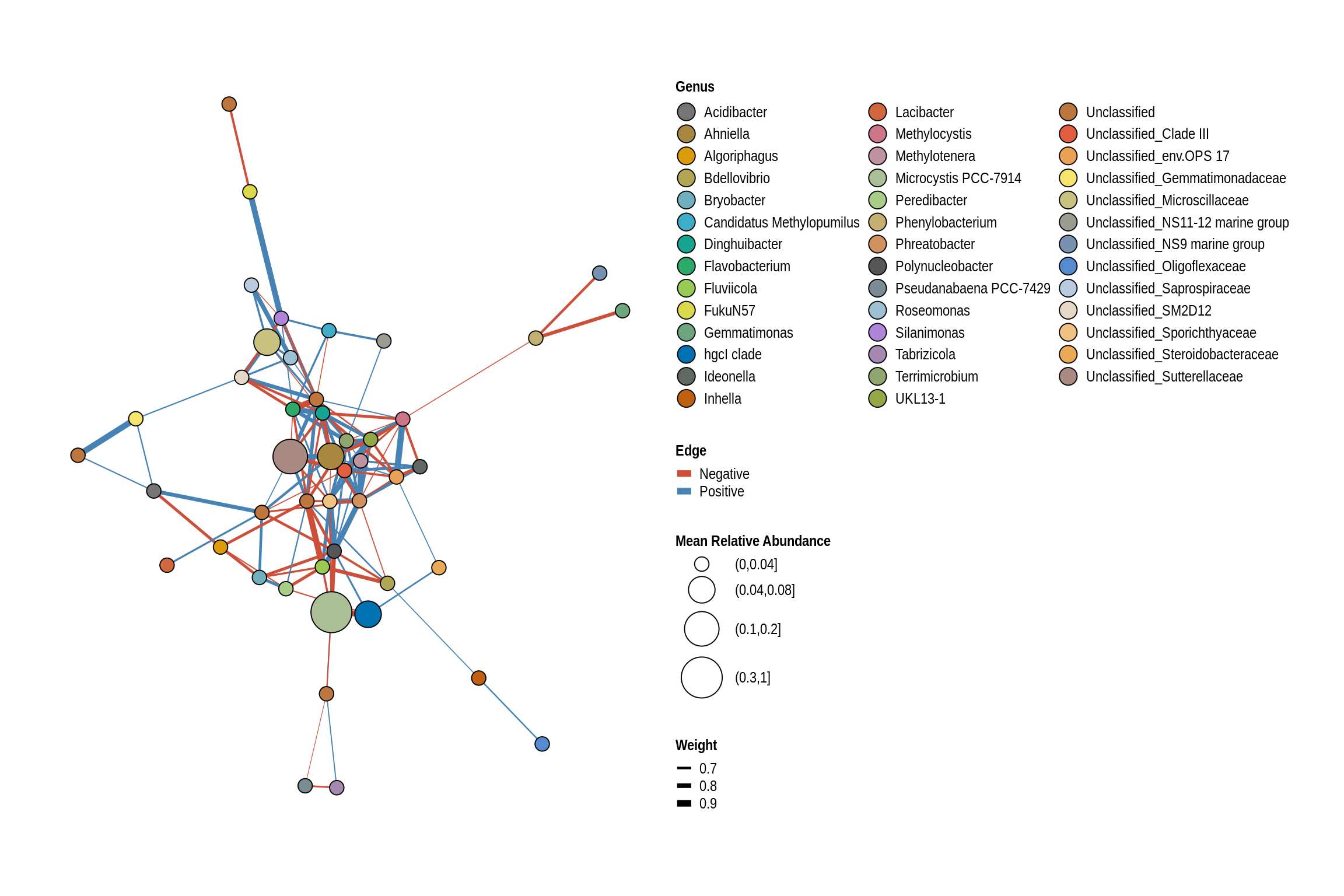
Lake Darling Beach
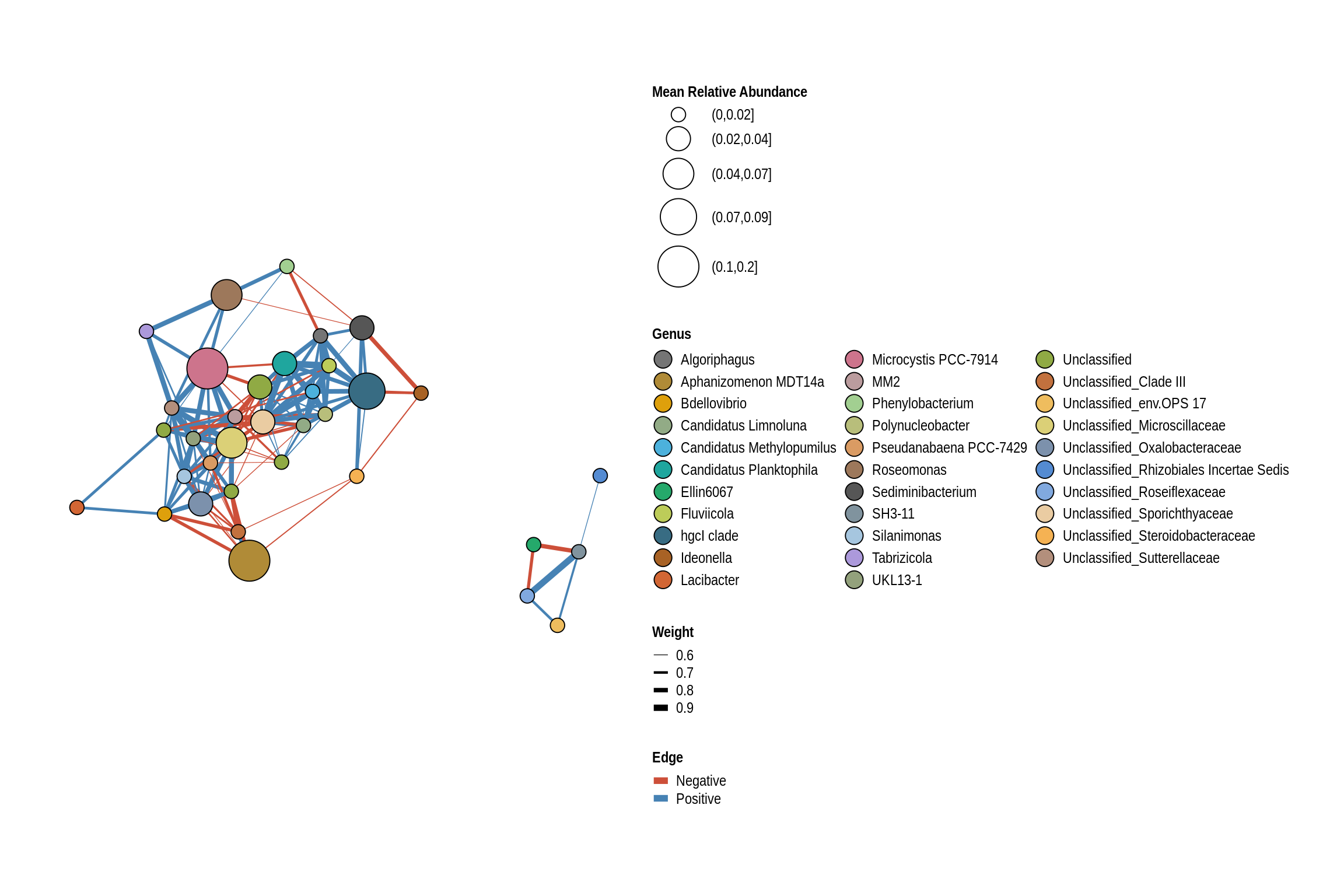
Brushy Creek Beach

Summary of Networks
# high_ <- high[!(Edge %in% low$Edge),]
high <- high[, .(Count = .N, rho = mean(rho)), by = c("X", "Y", "Edge")]
high <- high[Count >= 7]
setorder(high, -Count)
# low_ <- low[!(Edge %in% high$Edge),]
low <- low[, .(Count = .N, rho = mean(rho)), by = c("X", "Y", "Edge")]
low <- low[Count >= 6]
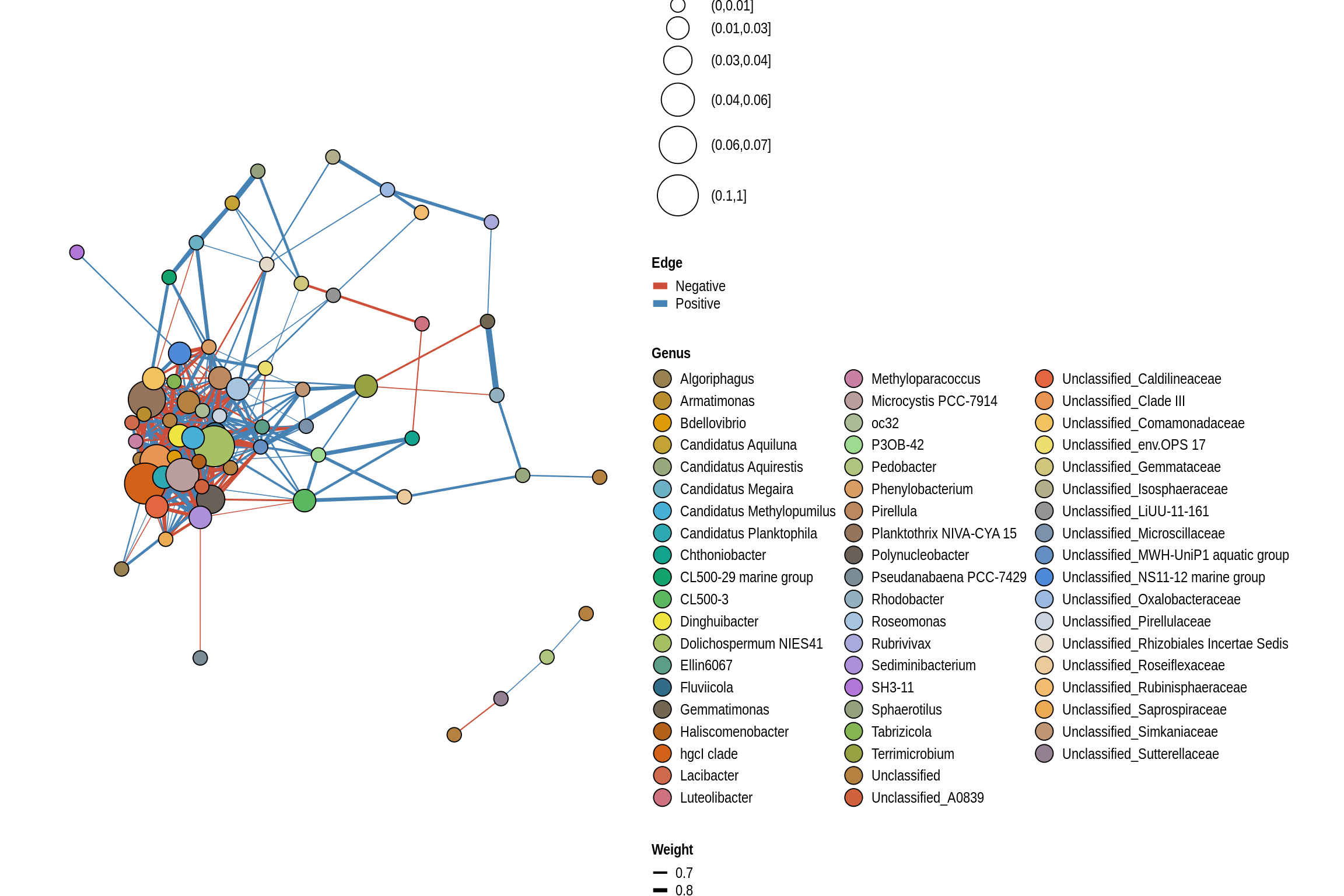
setorder(low, -Count)Summary of Low-Risk Lakes
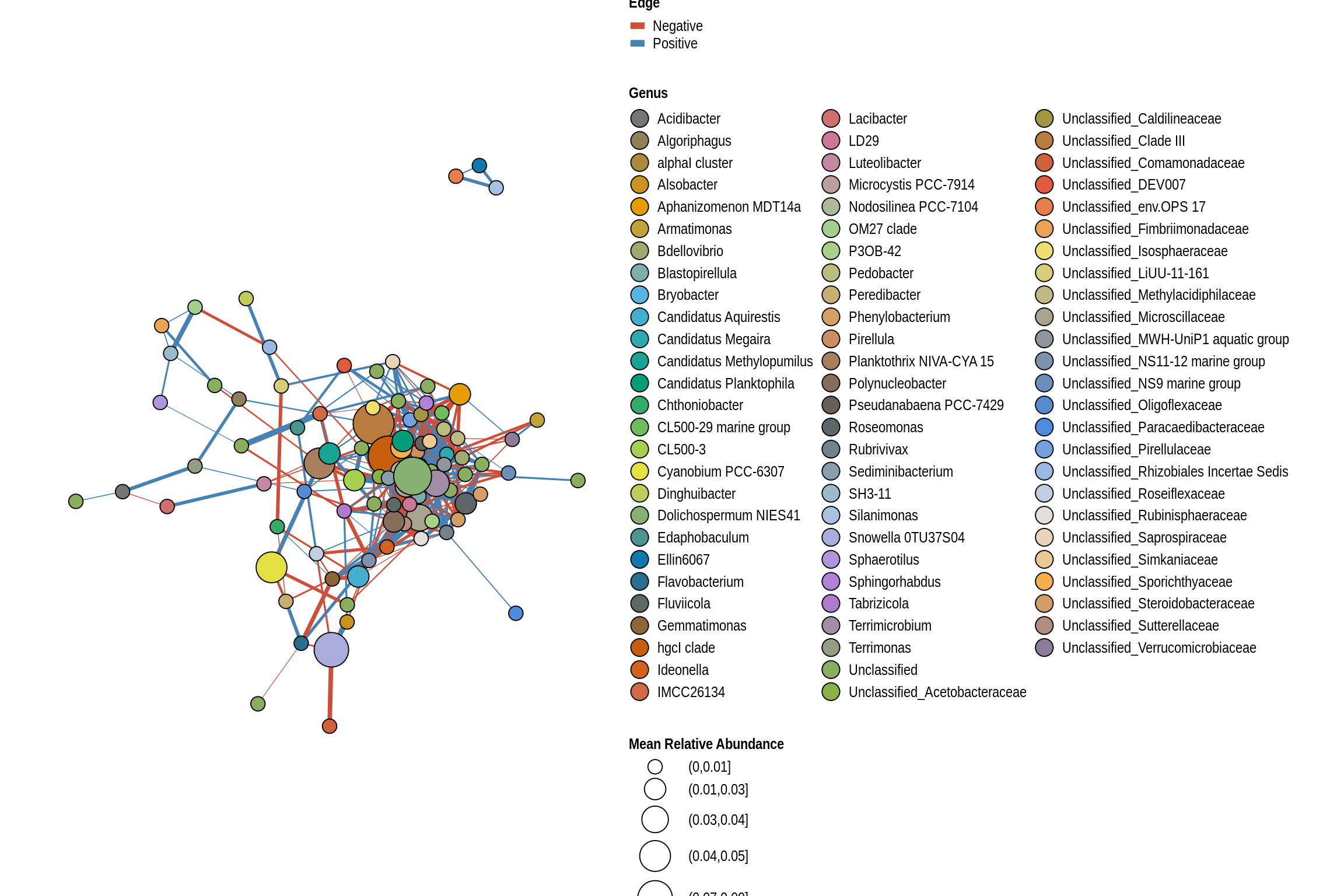
lowSummary of High-Risk Lakes
highco_occurrence_table <- high
phyloseq_obj <- relative_abundance(conglomerate_taxa(lake_hl, "Genus", TRUE, FALSE))
nodes <- data.table(as(access(phyloseq_obj, "tax_table"), "matrix"), keep.rownames = "Node_Name")
nodes[, `Mean Relative Abundance` := phylosmith:::bin(taxa_sums(phyloseq_obj)/nsamples(phyloseq_obj), nbins = 9)]
nodes <- nodes[nodes[["Node_Name"]] %in% c(as.character(co_occurrence_table$X),
as.character(co_occurrence_table$Y)), ]
co_occurrence_table[, `:=`(Weight, abs(rho))]
node_classes <- unique(c(co_occurrence_table$X, co_occurrence_table$Y))
clusters <- cluster_fast_greedy(simplify(graph_from_data_frame(d = co_occurrence_table, vertices = nodes, directed = FALSE), remove.multiple = TRUE, remove.loops = TRUE))$membership
cluster_sizes <- table(clusters)
nodes <- nodes[clusters %in% names(cluster_sizes[cluster_sizes > 2])]
co_occurrence_table <- co_occurrence_table[X %in% nodes$Node_Name &
Y %in% nodes$Node_Name]
net <- graph_from_data_frame(d = co_occurrence_table, vertices = nodes, directed = FALSE)
layout <- ggraph::create_layout(net, layout = "igraph", algorithm = "fr")
cluster <- cluster_fast_greedy(simplify(net, remove.multiple = TRUE, remove.loops = TRUE))$membership
communities <- data.table(layout[, c(1, 2)])
circles <- as(sf::st_buffer(sf::st_as_sf(communities, coords = c("x", "y")), dist = 0.5, nQuadSegs = 15), "Spatial")
circle_coords <- data.frame()
for (i in seq(nrow(communities))) {
circle_coords <- rbind(circle_coords, circles@polygons[[i]]@Polygons[[1]]@coords)
}
colnames(circle_coords) <- c("x", "y")
communities[, `:=`("Community", factor(cluster, levels = sort(unique(cluster))))]
communities <- data.table(circle_coords, Community = unlist(lapply(communities$Community, rep, times = nrow(circles@polygons[[i]]@Polygons[[1]]@coords))))
communities <- communities[, .SD[chull(.SD)], by = "Community", ]
hulls <- communities[, .SD[chull(.SD)], by = "Community", ]
community_count = length(unique(cluster))
community_colors <- create_palette(community_count, "#979aaa")
taxa <- as.data.table(as(lake_po@tax_table, 'matrix'), keep.rownames = "ASV")
layout_tax <- data.table(layout)
setnames(layout_tax, "name", "Genus")
layout_tax[taxa, on = 'Genus', Phylum := i.Phylum]
# layout_tax <- layout_tax[Genus != "Unclassified"]
node_colors <- phylosmith:::create_palette(length(unique(Phyla)))
layout$Phylum <- factor(layout$Phylum, levels = c(Phyla, unique(layout$Phylum)[!unique(layout$Phylum) %in% Phyla]))
# node_colors <- node_colors[factor(layout_tax$Phylum, levels = Phyla)]
# layout <- layout[!is.na(layout$Phylum),]classification <- "Phylum"
g <- ggraph(layout) + theme_graph() + coord_fixed()
g <- g + geom_polygon(data = hulls, aes_string(x = "x", y = "y", group = "Community"),
color = "#414a4c",
alpha = 0.2,
fill="#979aaa")
g <- g + geom_edge_link(aes_string(width = 'Count'), color = "tomato3") +
scale_edge_width_continuous(range = c(0.4, 2)) +
guides(colour = FALSE, alpha = FALSE, fill = guide_legend(ncol = 1), override.aes = list(size = 4))
g <- g + geom_point(aes_string(x = "x", y = "y", fill = "Phylum",
size = "`Mean Relative Abundance`"), pch = 21, color = "black") +
scale_fill_manual(values = node_colors, aesthetics = "fill") +
scale_size_discrete(range = c(4, 12))
g <- g + theme(legend.text = element_text(size = 12),
legend.title = element_text(size = 12, face = "bold"),
legend.key.size = unit(4, "mm"), legend.spacing.x = unit(0.005, "npc")) +
guides(colour = guide_legend(override.aes = list(size = 10)),
alpha = FALSE, fill = guide_legend(ncol = ceiling(length(unique(layout[[classification]]))/12),
override.aes = list(size = 5)), edge_color = guide_legend(override.aes = list(edge_width = 2)))
nodes_of_interest <- c("Actinobac")
nodes_of_interest <- c("Cyanobacteria", "Roseo", "Phenyl", "Ideon", "Sphaero", "Limnohabitans", "Limnoluna",
"Unclassified_Microscillaceae", "CL500", "GKS98", "hgc", "Silan")
coi <- subset(layout, apply(layout, 1, function(class) {
any(grepl(paste(nodes_of_interest, collapse = "|"),
class))
}))
g <- g + ggrepel::geom_label_repel(data = coi,
aes_string(x = "x", y = "y", fill = classification),
box.padding = unit(2, "lines"),
point.padding = unit(0.5, "lines"),
size = 4,
# arrow = arrow(length = unit(0.22, "inches"), ends = "last", type = "closed"),
show.legend = FALSE,
label = lapply(unname(apply(coi,
1, function(class) {
class[any(grepl(paste(nodes_of_interest,
collapse = "|"), class))][9]
})), `[[`, 1),
max.overlaps = 1000)
g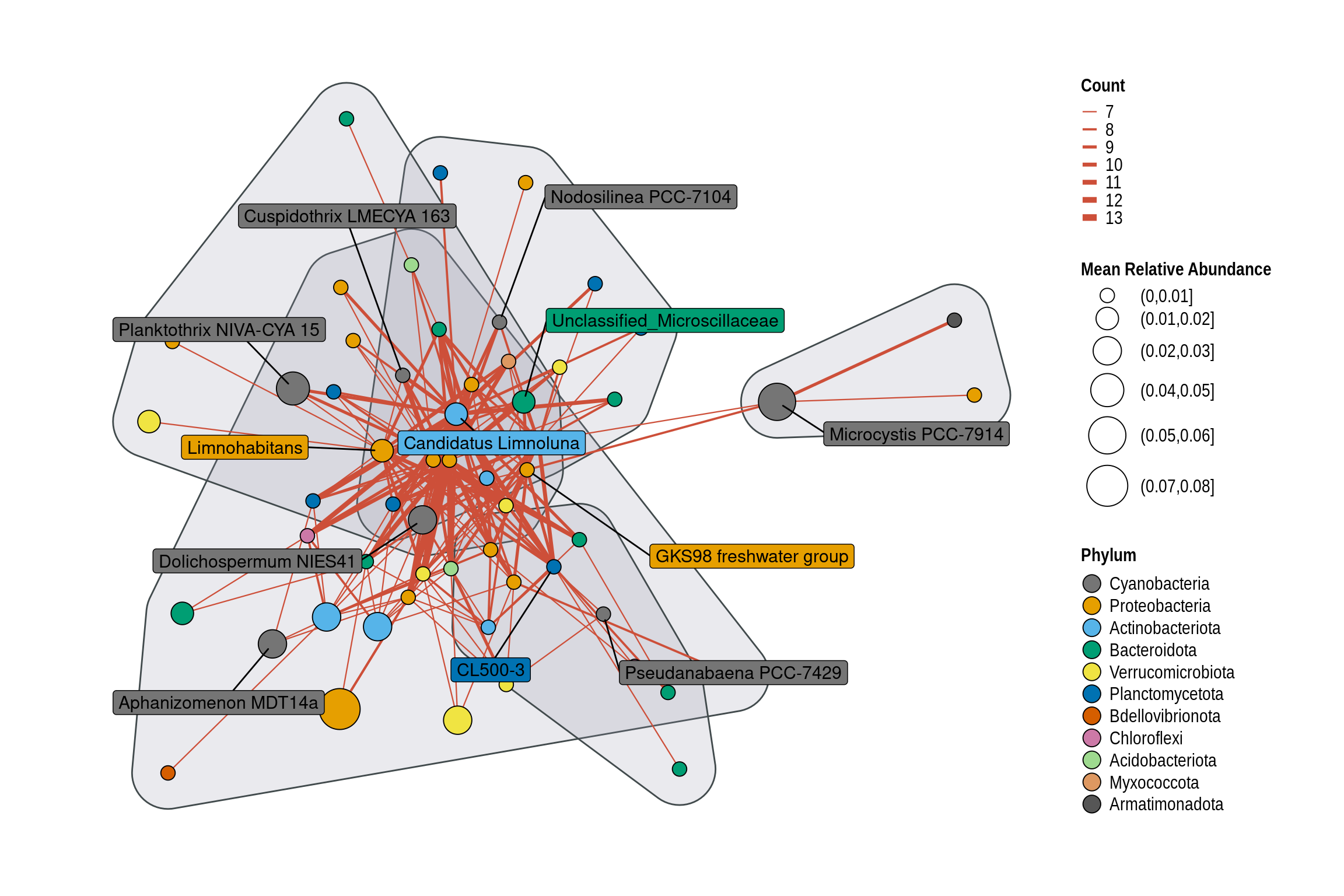
Schuyler Smith
Ph.D. Student - Bioinformatics and Computational Biology
Iowa State University. Ames, IA.