Primer Success Tables
BLAST_file_path = "../blast/"
if (length(BLAST_file_path) == 1 && dir.exists(BLAST_file_path)) {
BLAST_file_path <- gsub("/$", "", BLAST_file_path)
BLAST_file <- dir(BLAST_file_path, full.names = TRUE)
} else {
BLAST_file <- BLAST_file_path
}
BLAST_file <- sort(normalizePath(BLAST_file))
primer_success <- data.table::data.table(sample = character(),
target = character(),
n_reads = numeric(),
artifacts = numeric(),
analyzed = numeric(),
true_pos = numeric(),
false_pos = numeric(),
false_neg = numeric(),
true_neg = numeric())
for(file in BLAST_file){
sample_name <- gsub('Q3-','',basename(file))
files <- dir(file, full.names = TRUE)
files <- sort(normalizePath(files))
unknown <- files[grep('unknown', files)]
unknown <- data.table::fread(unknown, header = FALSE, sep = '\t',
col.names = c('query', 'reference', 'perc_id', 'length',
'mismatch', 'gap', 'qstart', 'qend',
'rstart', 'rend', 'e', 'bit'))
unknown <- unknown[length >= 90]
setorder(unknown, 'query', 'perc_id')
unknown <- merge(classifications, unknown, by.x = "Target", by.y = "reference")
unknown <- unique(unknown, by=c("query", "Target"))
unknown <- unknown[unknown[, .I[length == max(length, na.rm=T)], by = c('query', 'Target')][[3]]]
unknown <- unknown[unknown[, .I[perc_id == max(perc_id, na.rm=T)], by = c('query', 'Target')][[3]]]
unknown <- unknown[, .(count = .N), by = Target]
for(primer_file in files){
if(file.info(primer_file)$size > 0){
blast <- data.table::fread(primer_file, header = FALSE, sep = '\t')
#'query', 'reference', 'perc_id', 'length', 'mismatch', 'gap',
#'qstart', 'qend', 'rstart', 'rend', 'e', 'bit'
setorder(blast, 'V1', 'V3')
blast <- blast[V3 > 95]
blast <- merge(classifications, blast, by.x = "Target", by.y = "V2")
n_reads <- length(unique(blast[['V1']]))
blast <- blast[V4 >= 90]
analyzed <- length(unique(blast[['V1']]))
n_artifacts <- n_reads - analyzed
primer_target <- gsub('____.?', '', basename(sapply(strsplit(primer_file, "\\."), "[[", 1)))
false_neg <- unknown[Target == primer_target][[2]]
successes <- 0
if(nrow(blast) > 0){
blast <- blast[blast[, .I[V4 == max(V4, na.rm=T)], by = c('V1', 'Target')][[3]]]
blast <- unique(blast, by=c("V1", "Target"))
blast <- blast[, .(count = .N), by = Target]
successes <- blast[Target == primer_target][[2]]
}
if(length(successes) < 1) successes <- 0
if(length(false_neg) < 1) false_neg <- 0
primer_success <- rbind(primer_success,
data.table::data.table(
sample = sample_name,
target = primer_target,
n_reads = n_reads,
artifacts = n_artifacts,
analyzed = analyzed,
true_pos = successes,
false_pos = (analyzed - successes),
true_neg = 0,
false_neg = false_neg))
}
}
set(primer_success, primer_success[, .I[sample == sample_name]], 'true_neg', sum())
print(sample_name)
}
primer_success[,true_neg := sum(true_pos)-true_pos, by = sample]
primer_success <- merge(classifications, primer_success, by.x = 'Target', by.y = 'target')
primer_success <- primer_success[n_reads != 'NA']
primer_success <- primer_success[ARG_Family != '16S']
saveRDS(primer_success, '../data/primers/primer_success.RDS')
data.table::set(primer_success, which(primer_success[["success_rate"]] == 0), "success_rate", NA)
primer_success[, experiment := sapply(strsplit(sample, "-"), "[[", 1)]
read_counts <- data.table::dcast(primer_success, Target ~ sample, value.var=c("true_pos"), fun = sum)
read_counts <- read_counts[c(which(rowSums(read_counts[,-1]) > 0)),]
read_counts <- merge(classifications, read_counts, by = 'Target')
saveRDS(read_counts, '../data/primers/read_counts_from_primers.RDS')Overall for Primers
samples <- primer_success[, sum(true_pos, na.rm = T), by = sample][V1 > 1000]$sample
primer_success <- primer_success[sample %in% samples]
primers <- primer_success[, lapply(.SD, sum, na.rm=TRUE), by=Target, .SDcols=c("n_reads", "artifacts", "analyzed", "true_pos", "false_pos") ]
setorder(primers, -n_reads)
Overall for Samples
sample_success <- primer_success[, lapply(.SD, sum, na.rm=TRUE), by=sample, .SDcols=c("n_reads", "artifacts", "analyzed", "true_pos", "false_pos") ]
setorder(sample_success, sample)
True-Positive Reads by Samples
data.table::set(primer_success, which(primer_success[["success_rate"]] == 0), "success_rate", NA)
primer_success[, experiment := sapply(strsplit(sample, "-"), "[[", 1)]
read_counts <- data.table::dcast(primer_success, Target ~ sample, value.var=c("true_pos"), fun = sum)
read_counts <- read_counts[c(which(rowSums(read_counts[,-1]) > 0)),]
read_counts <- merge(classifications, read_counts, by = 'Target')
Success Metrics
primer_success[, experiment := NULL]
setorder(primer_success, -n_reads)primer_success[, sensitivity := round(true_pos/(true_pos + false_neg)*100, 1)]
primer_success[, specificity := round(true_neg/(true_neg + false_pos)*100, 1)]
primer_success[, accuracy := round(true_pos/(true_pos + false_neg)*100, 1)]
Mean Sensitivity
Mean sensitivity of each primer, weighted by abundance per sample, was found to be 99.56
Mean Specificity
Mean specificity of each primer, weighted by abundance per sample, was found to be 99.92
Mean Accuracy
Mean accuracy of each primer, weighted by abundance per sample, was found to be 99.56
Primer Performance Graphs
primers <- primer_success[, lapply(.SD, sum, na.rm=TRUE), by=Target, .SDcols=c("n_reads", "artifacts", "analyzed", "true_pos", "false_pos", "false_neg", "true_neg") ]
primers[, percent_artifacts := artifacts/n_reads*100]
primers[, percent_tp := true_pos/n_reads*100]
primers[, percent_fp := false_pos/n_reads*100]
primers[, percent_fn := false_neg/n_reads*100]All
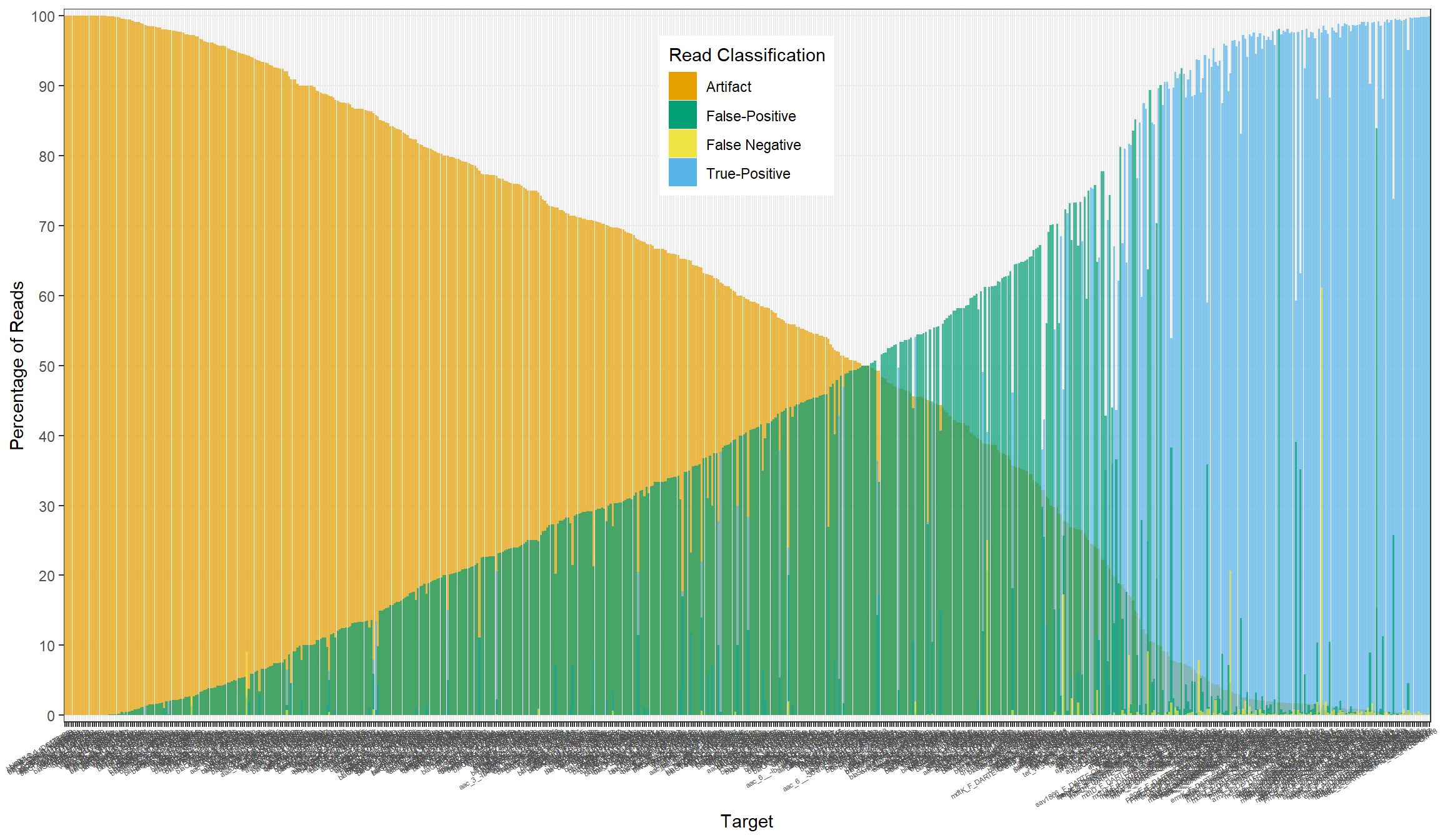
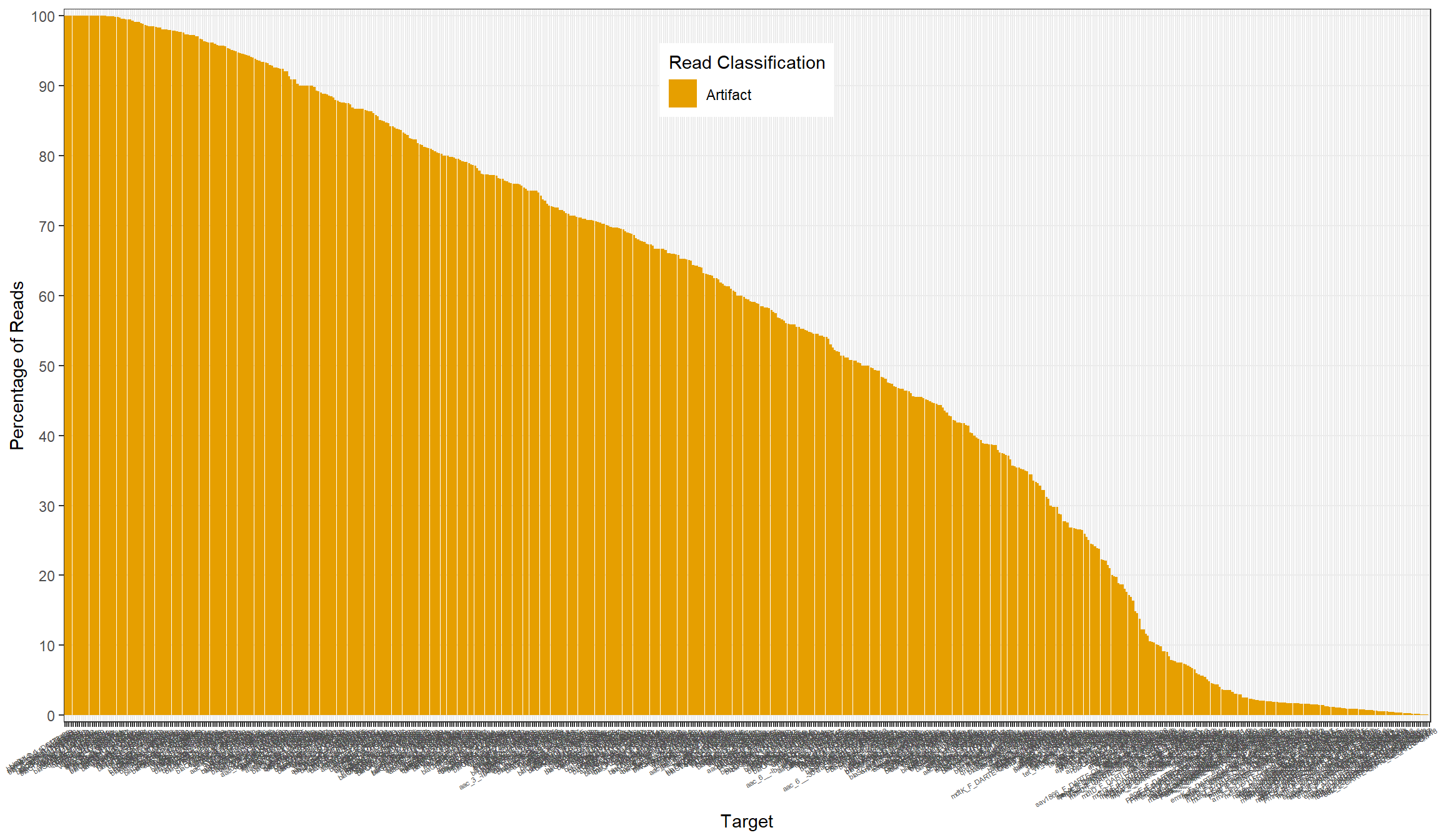
Artifact
True-Positive

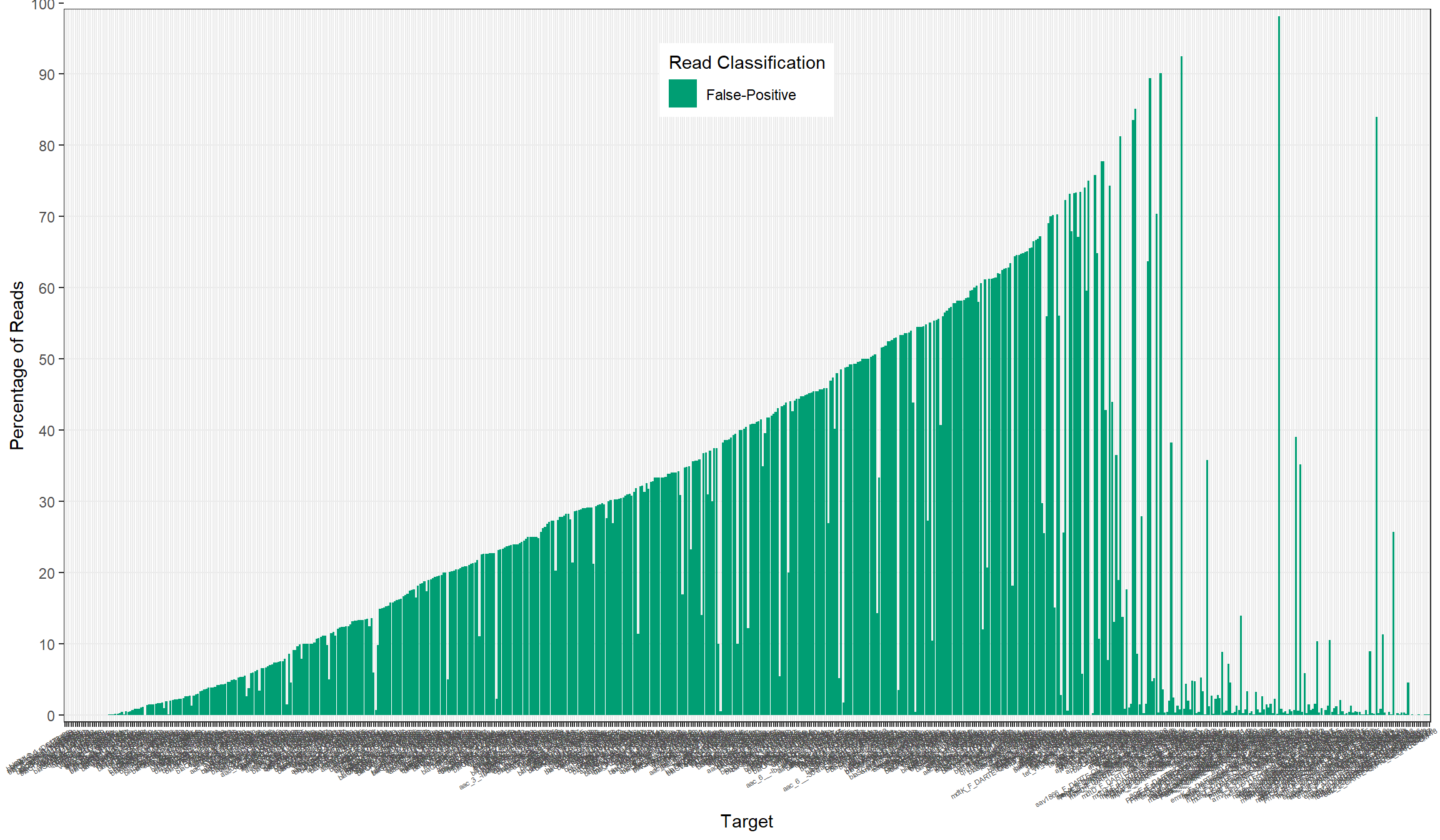
False-Positive
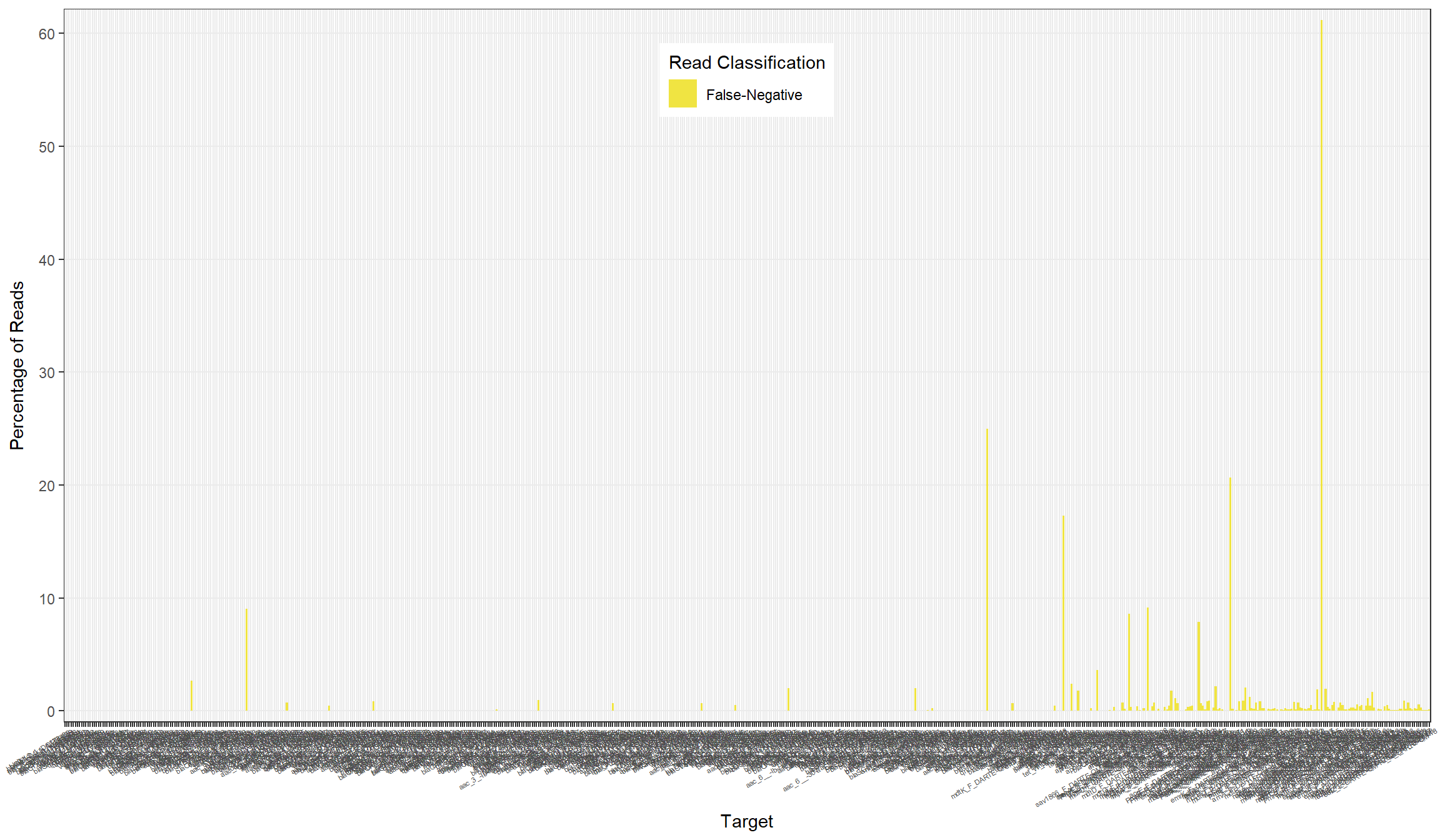
False-Negative
Artifact Reads
Examples of Artifact Reads
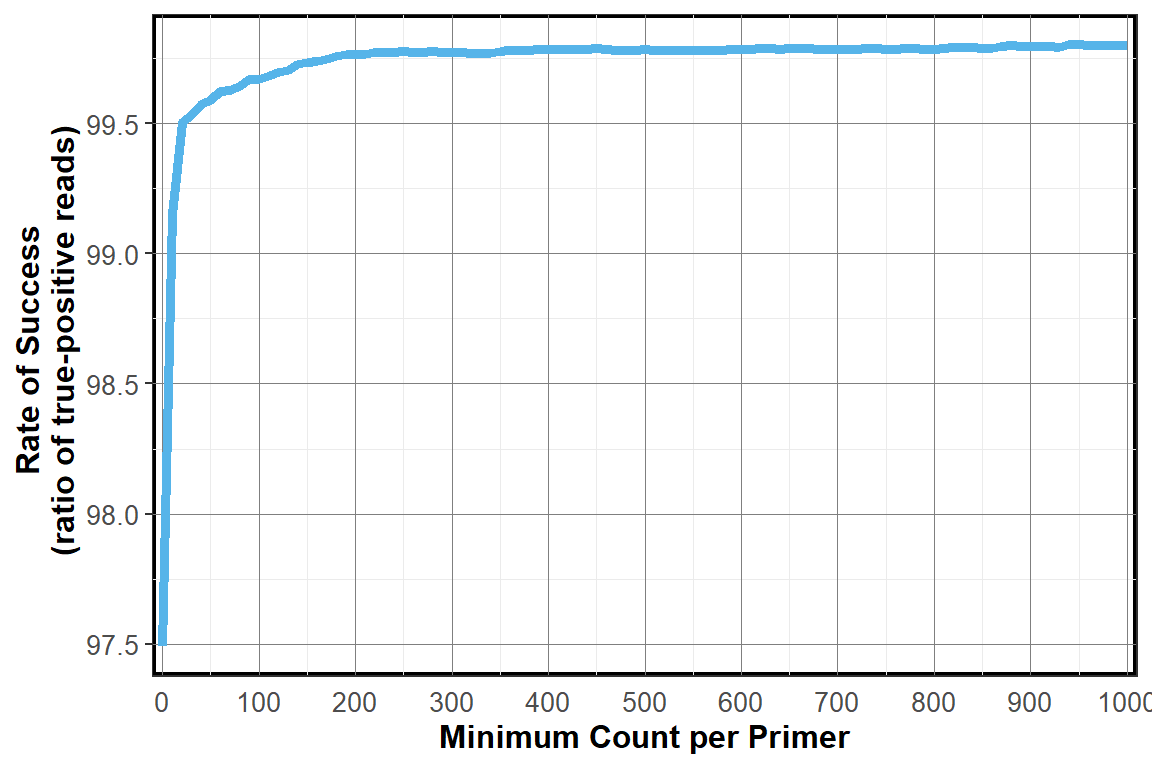
Sensitivity by True-Postive Count

True-Positives by Read Count

Artifacts by Primer Count
Correlation of Artifacts and Read Count
The number of reads produced per sample and the percentage of those reads that were PCR-artifacts had a negative correlation with an R-squared value = 0.73

Schuyler Smith
Ph.D. Student - Bioinformatics and Computational Biology
Iowa State University. Ames, IA.