Sequencing Results
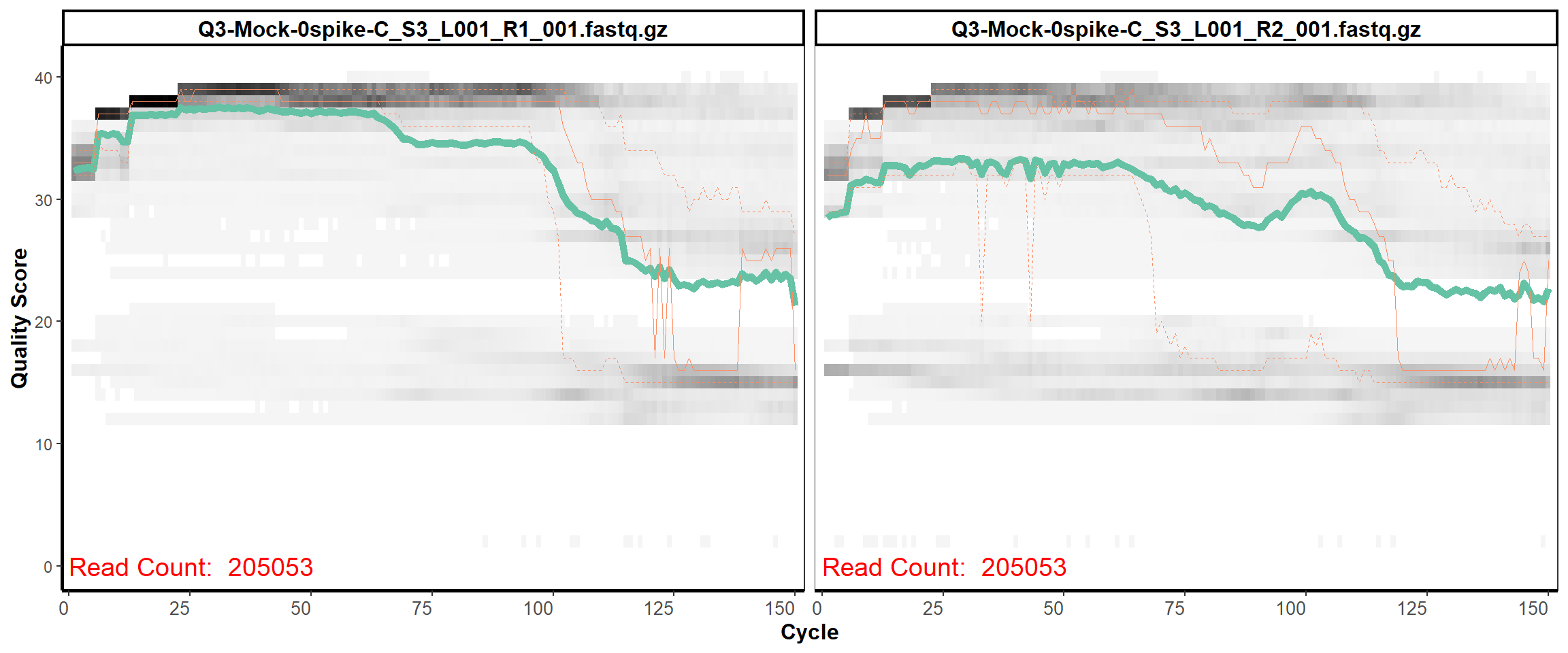
Mock-Community
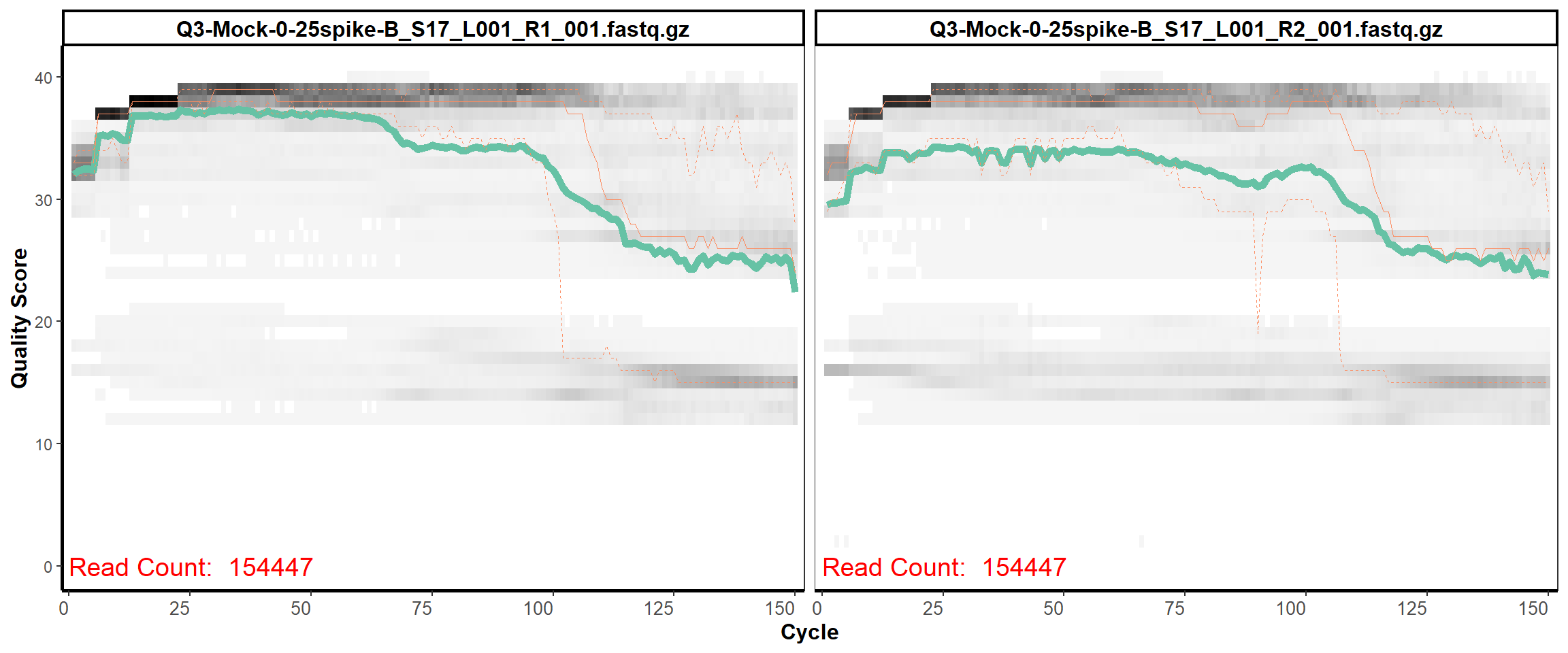
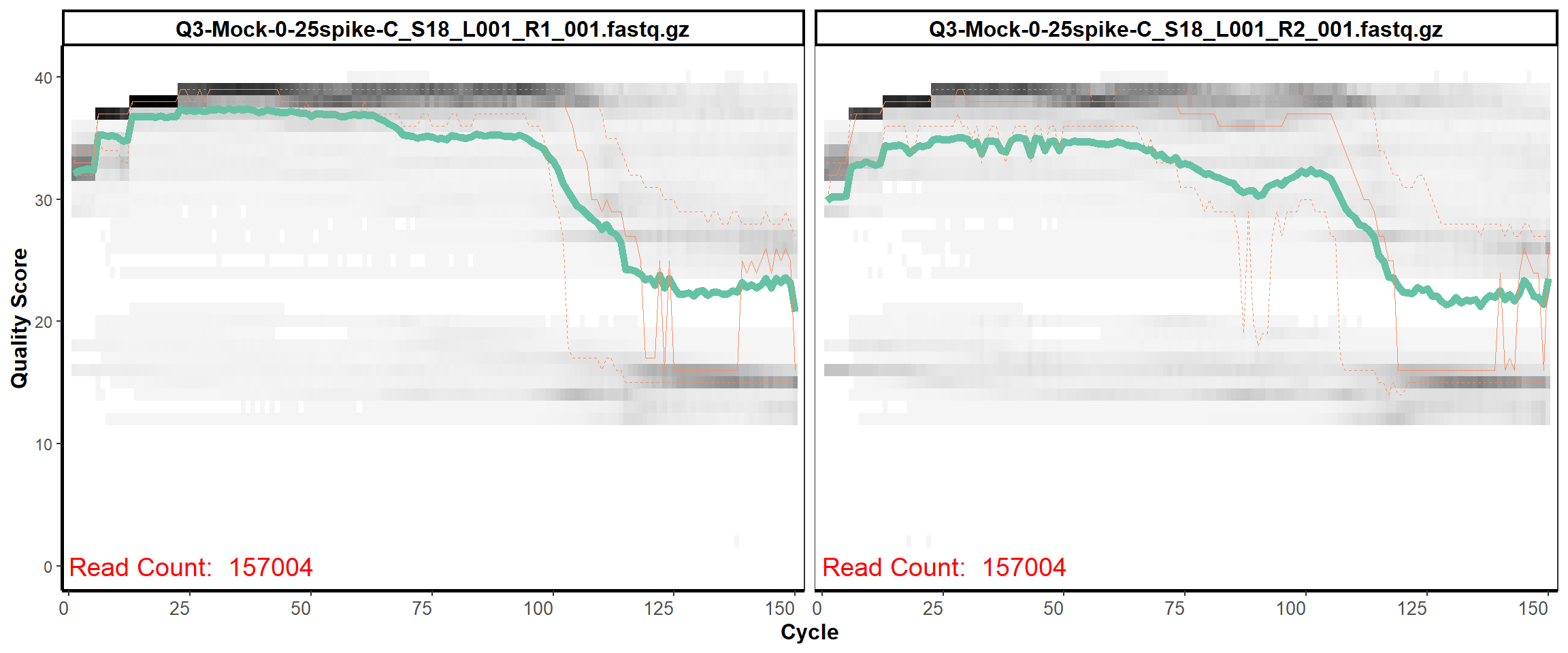
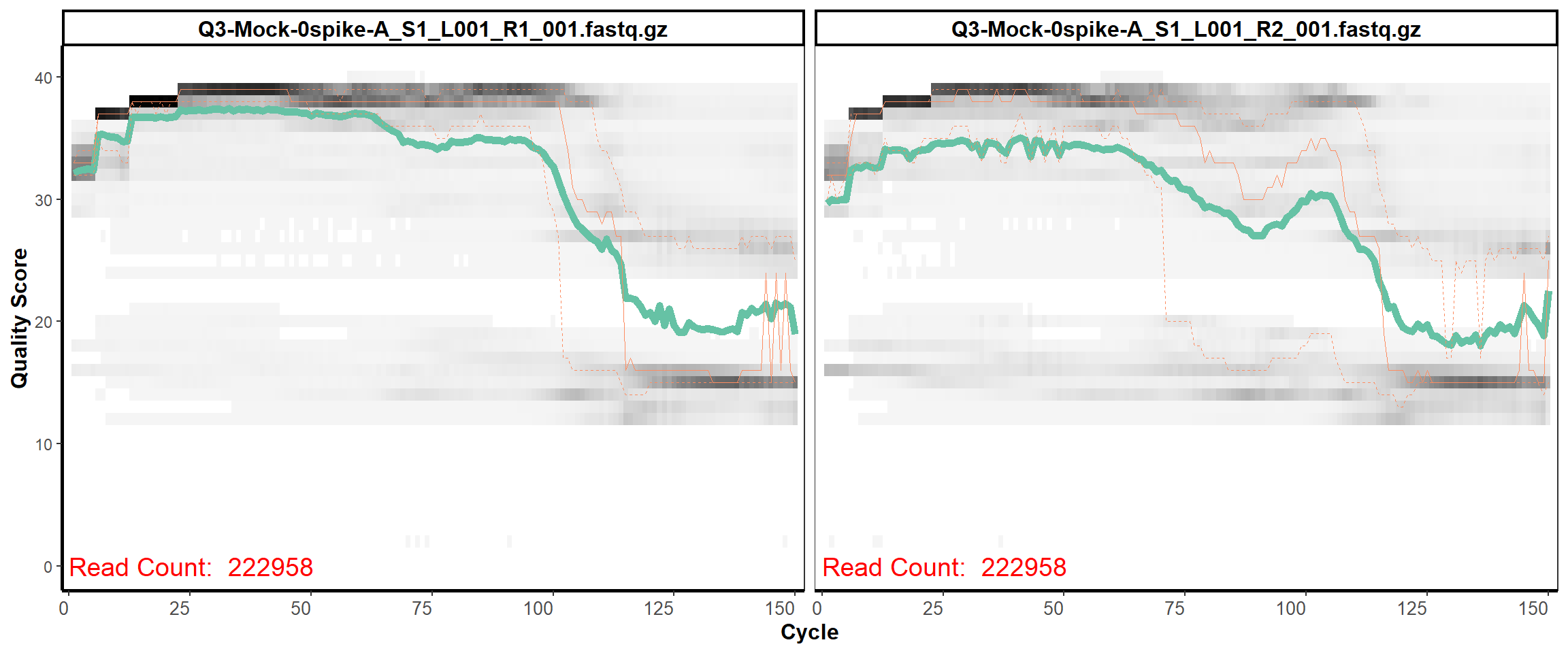
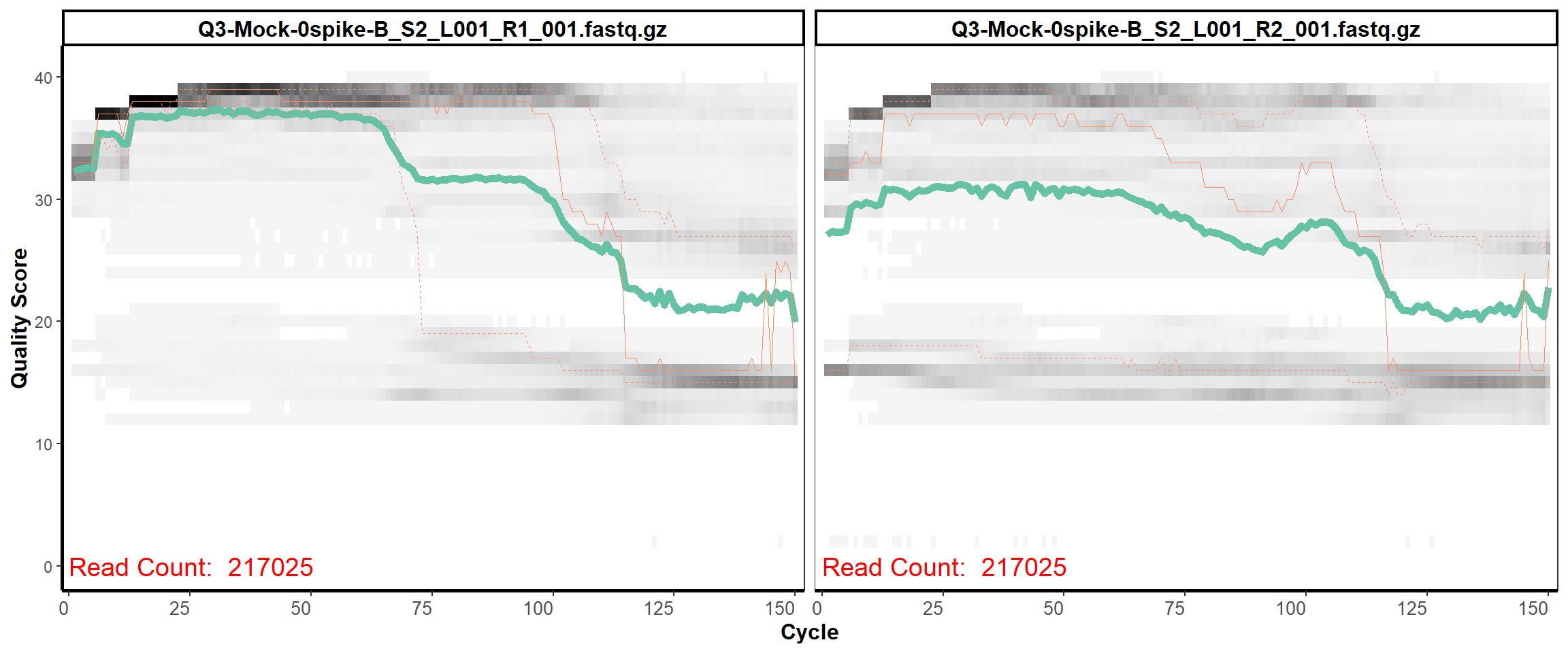
MiSeq Quality scores
1
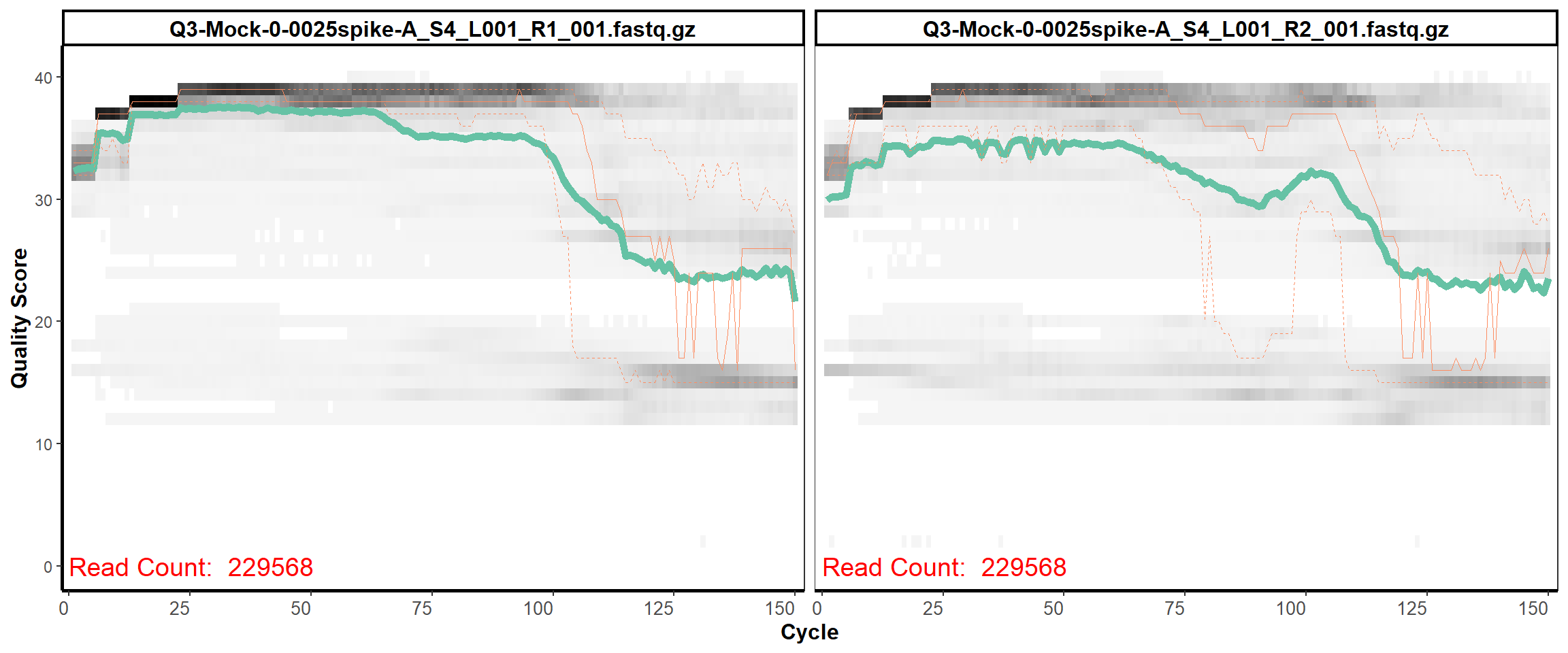
2
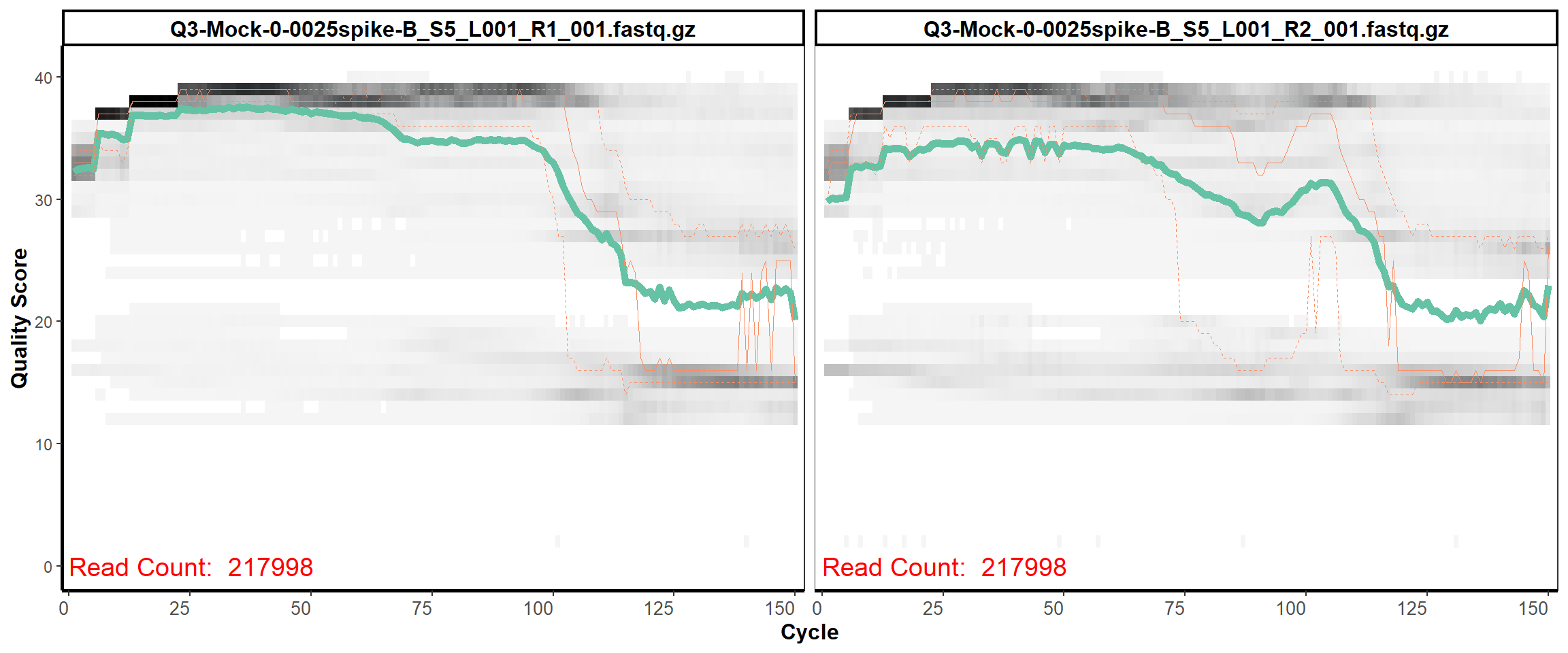
3
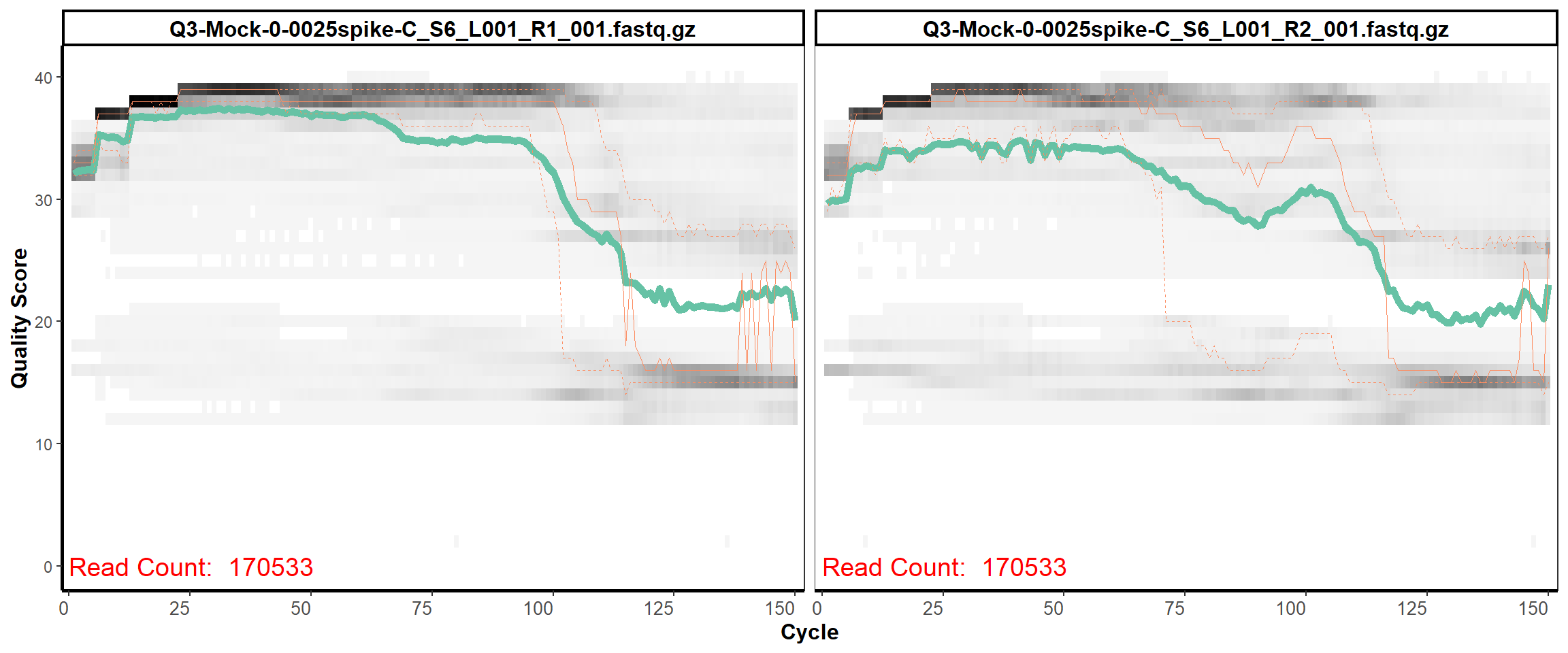
4
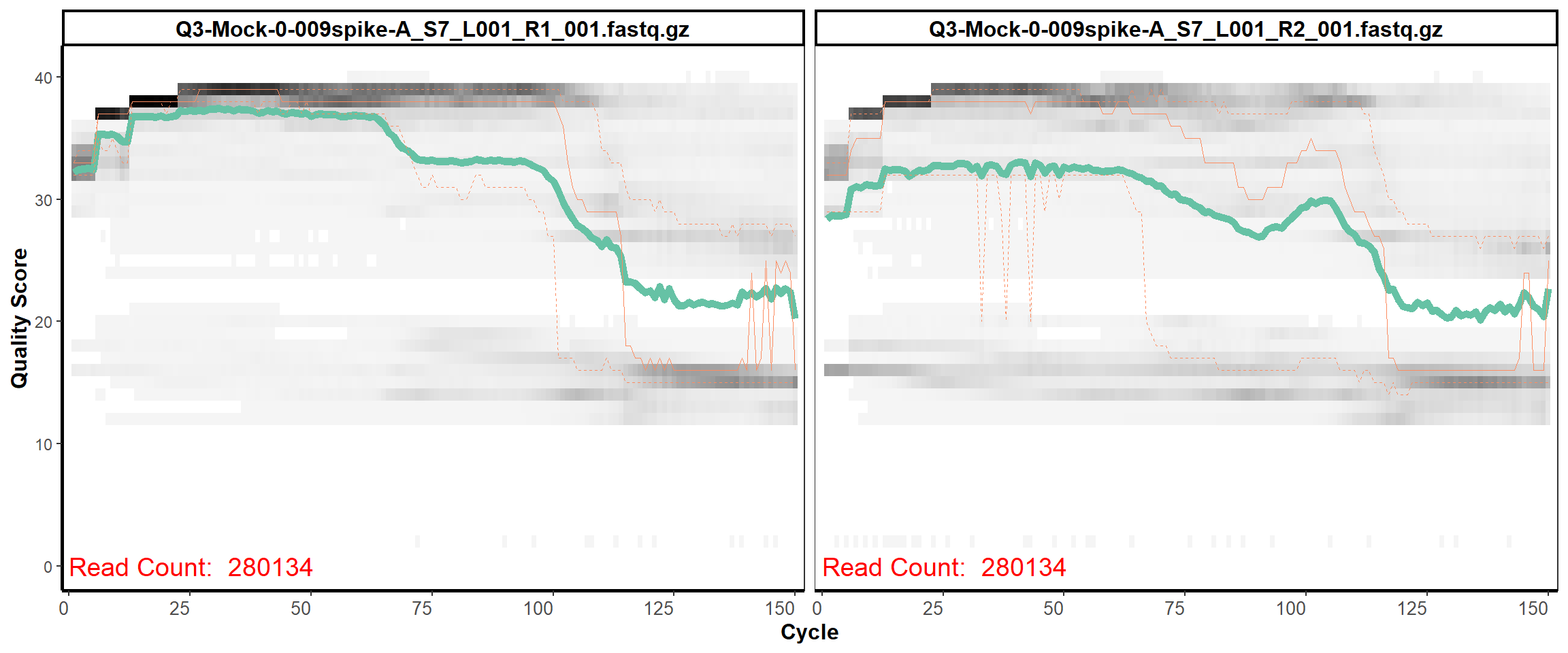
5
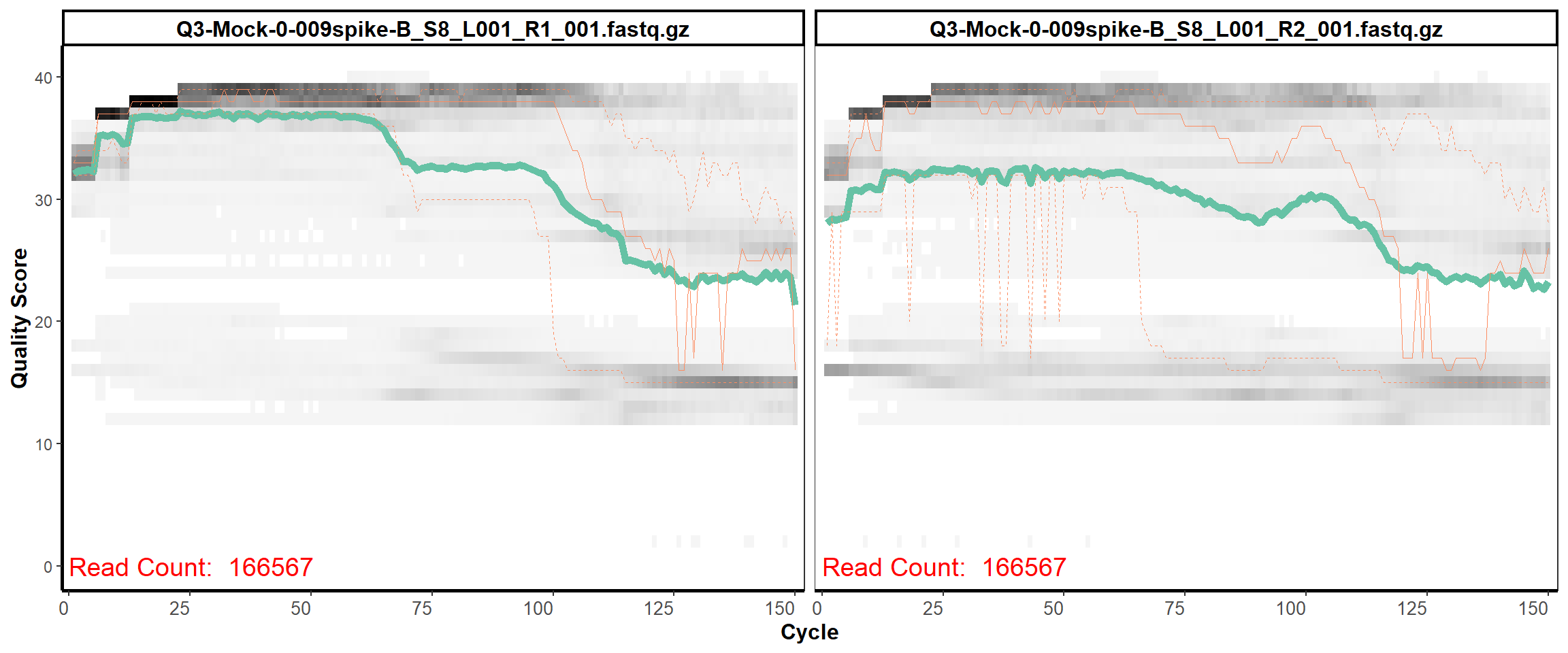
6
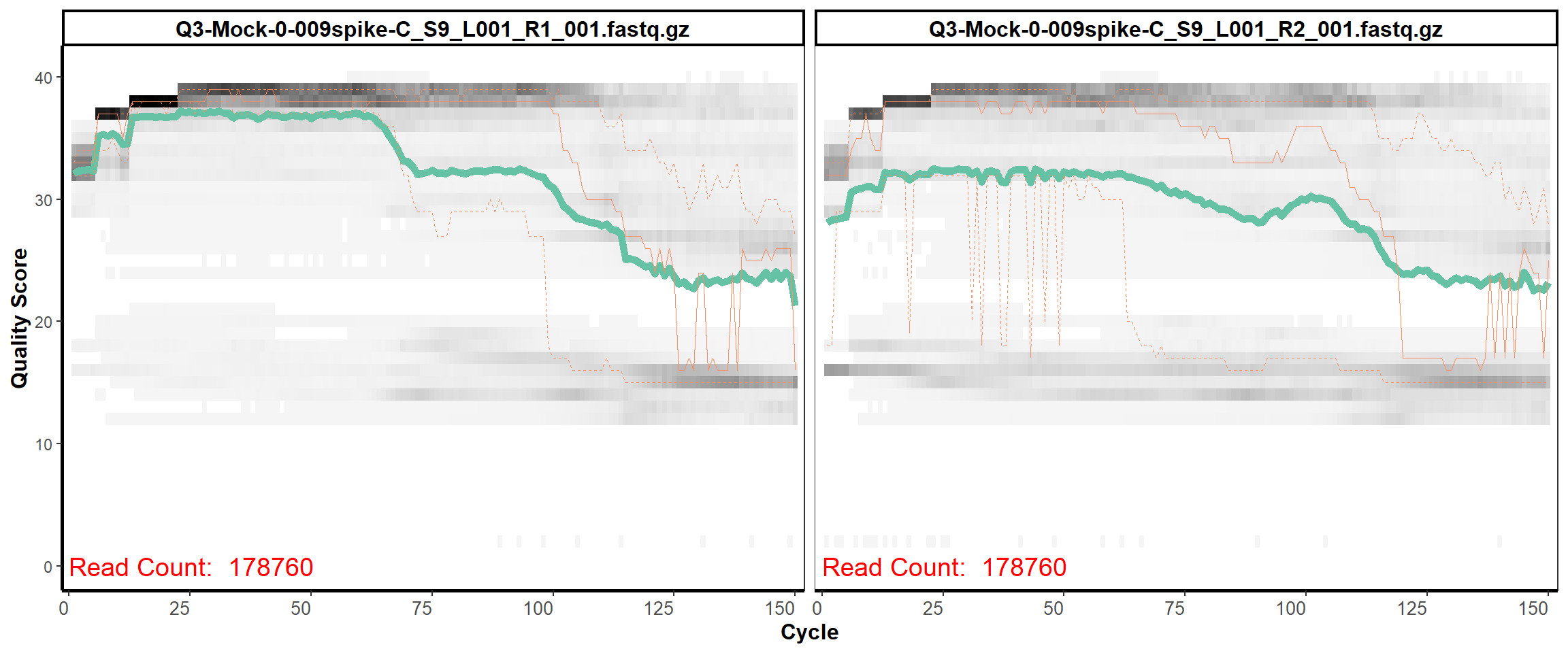
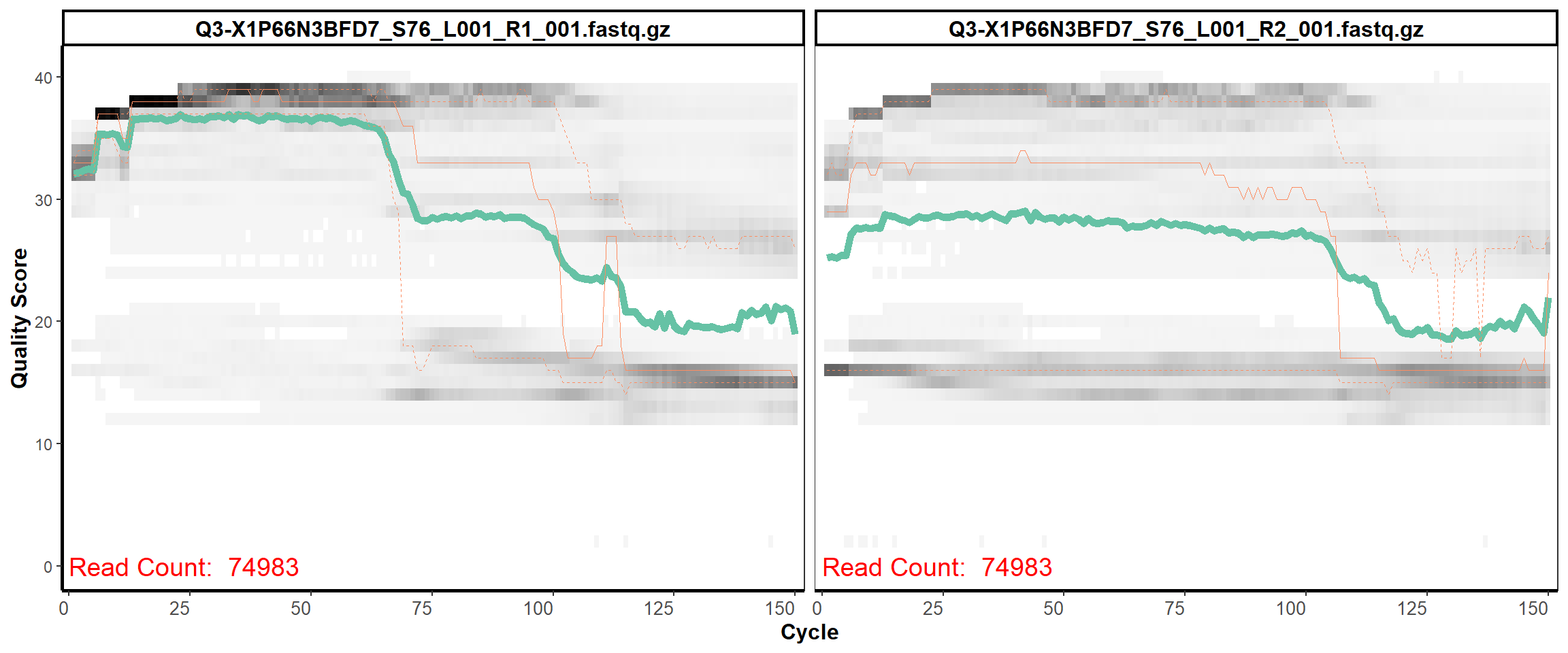
7

8
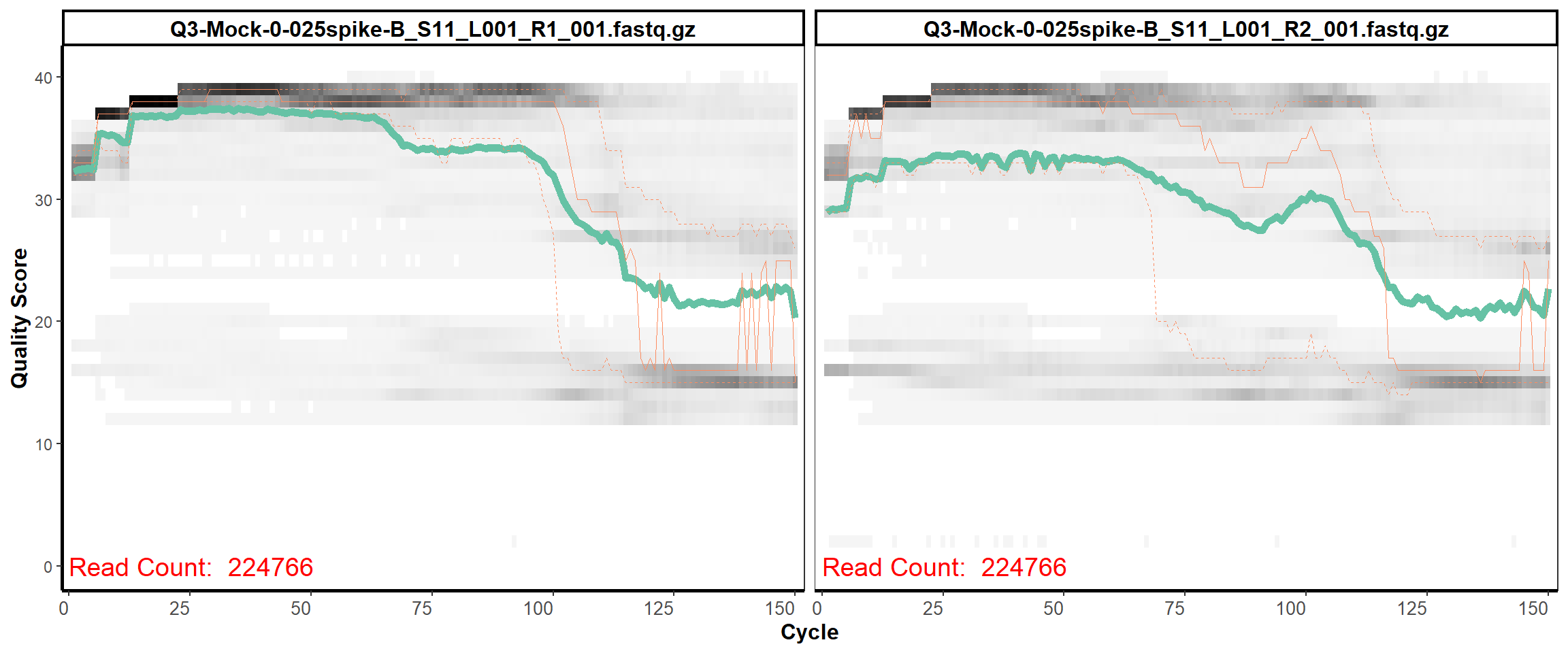
9
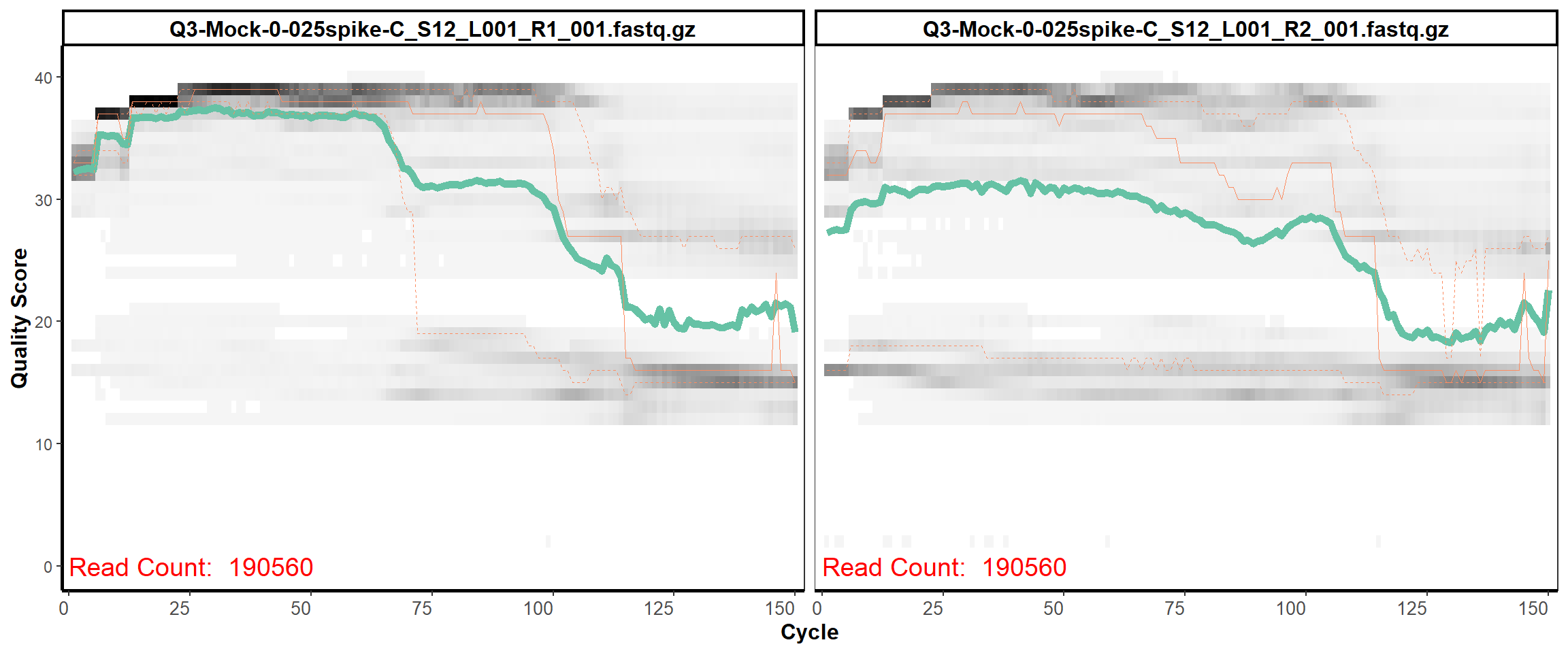
10
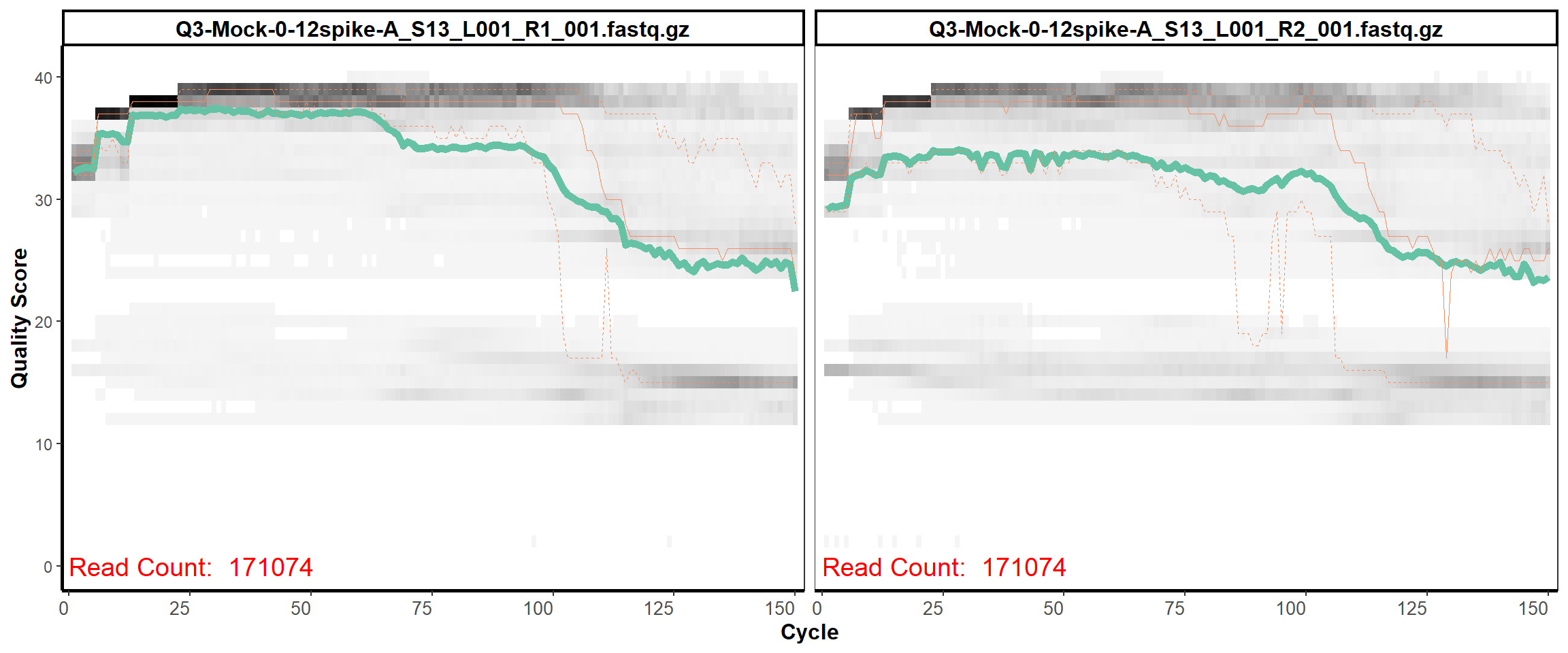
11
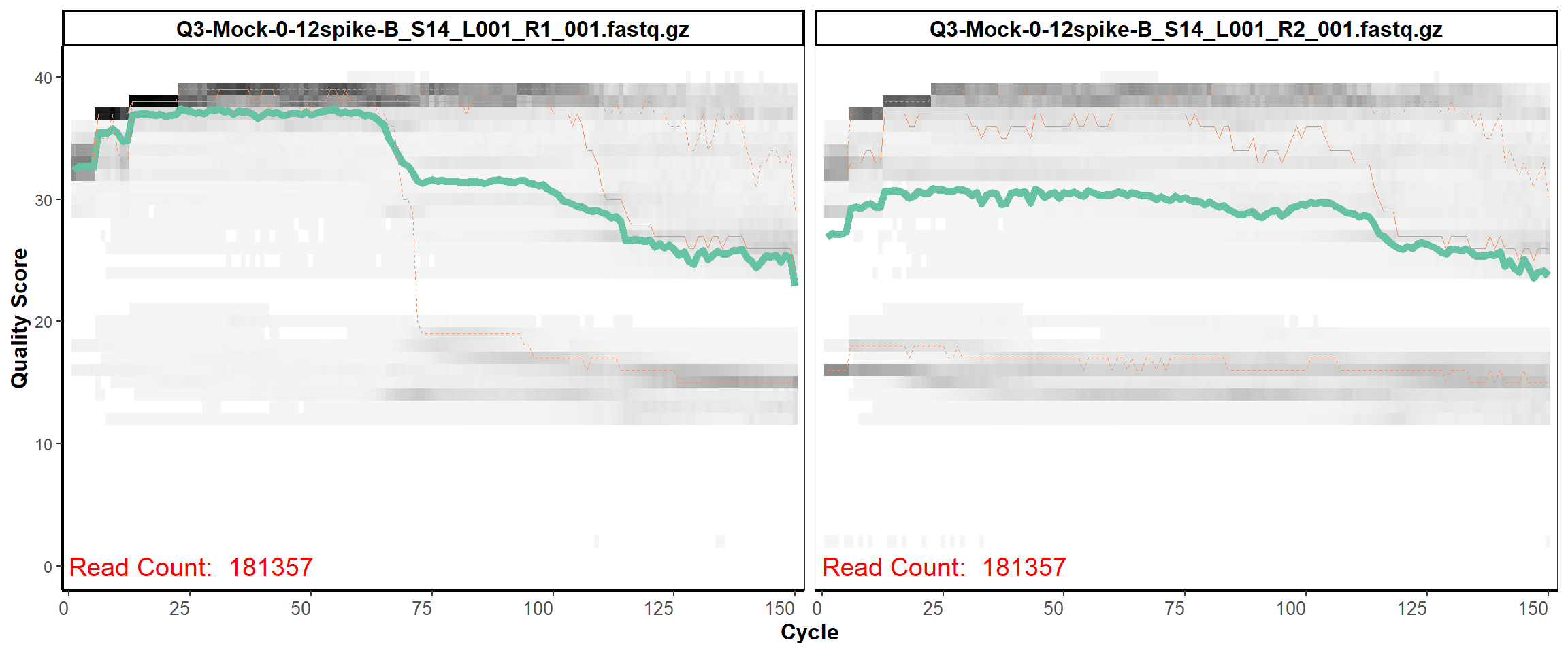
12
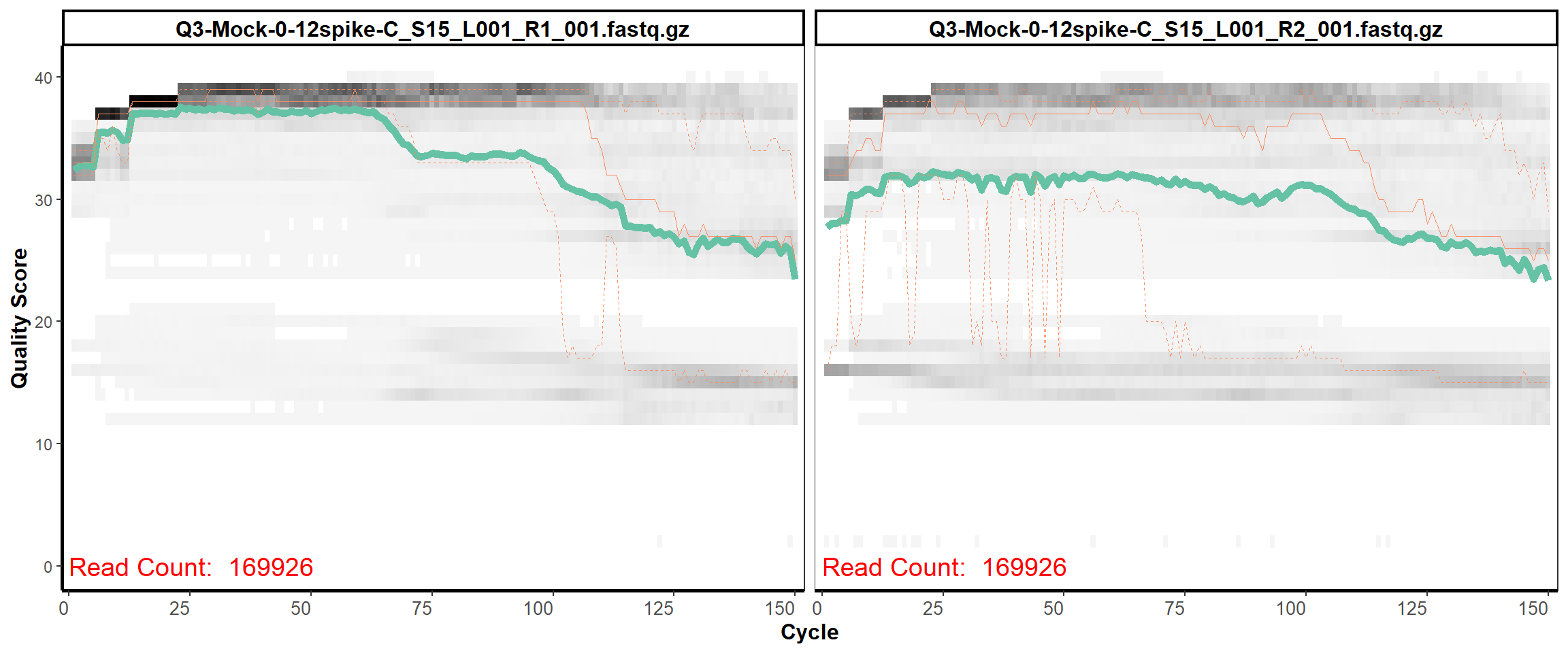
13
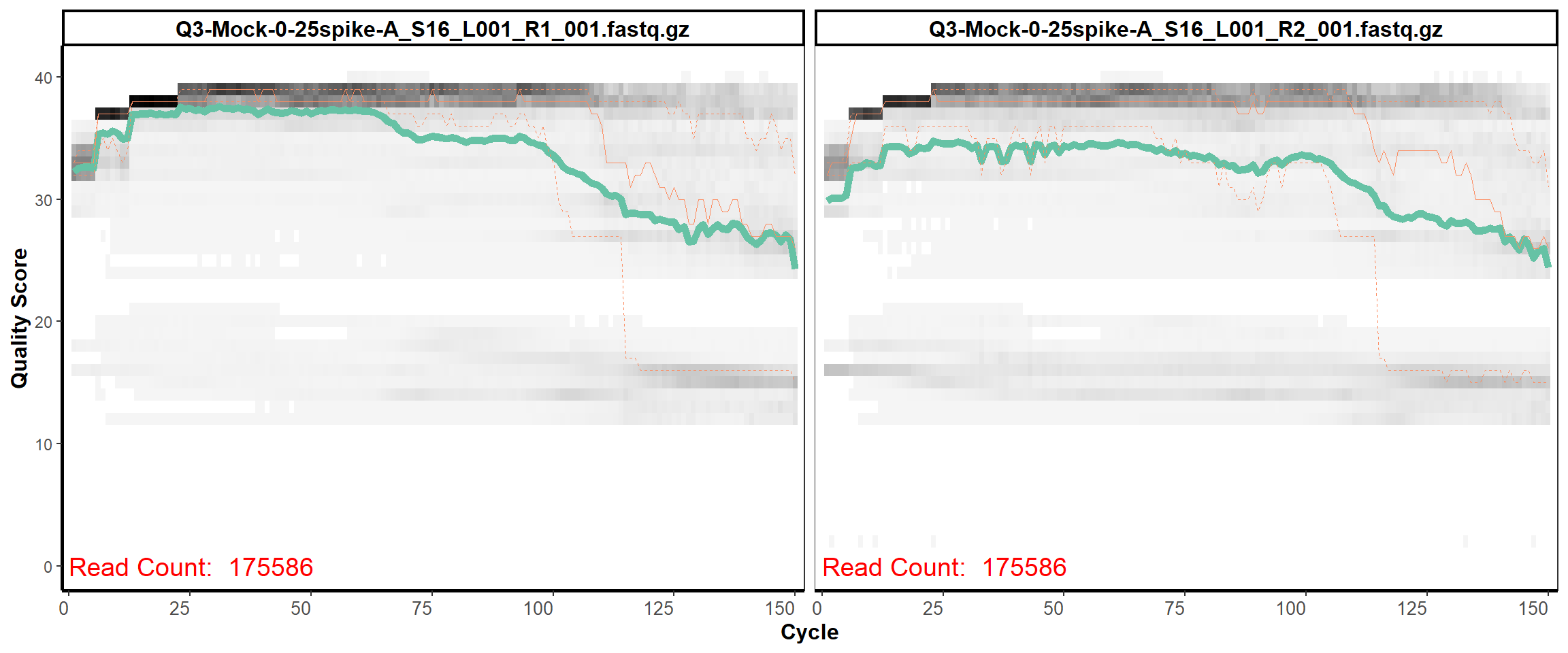
14
15
16
17
18
Sequence Results
Soil Column I
MiSeq Quality scores
1
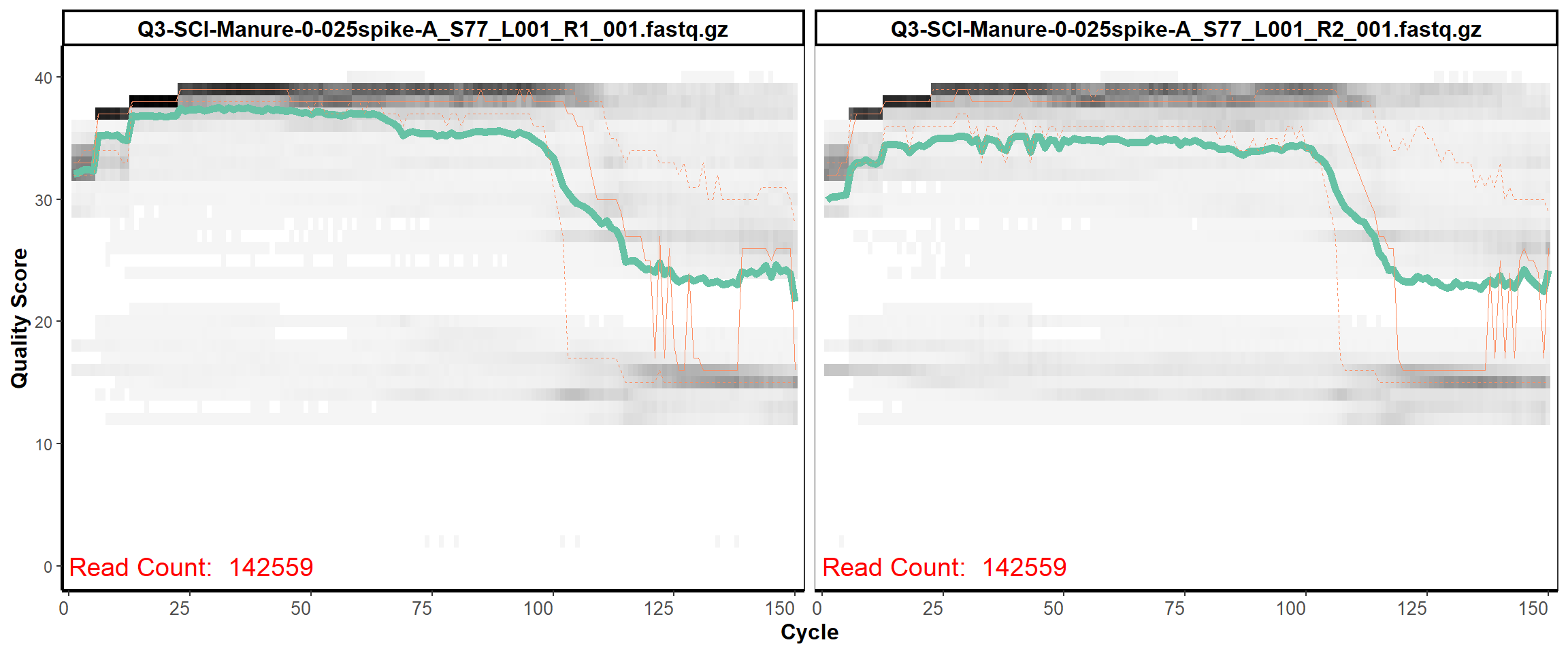
2
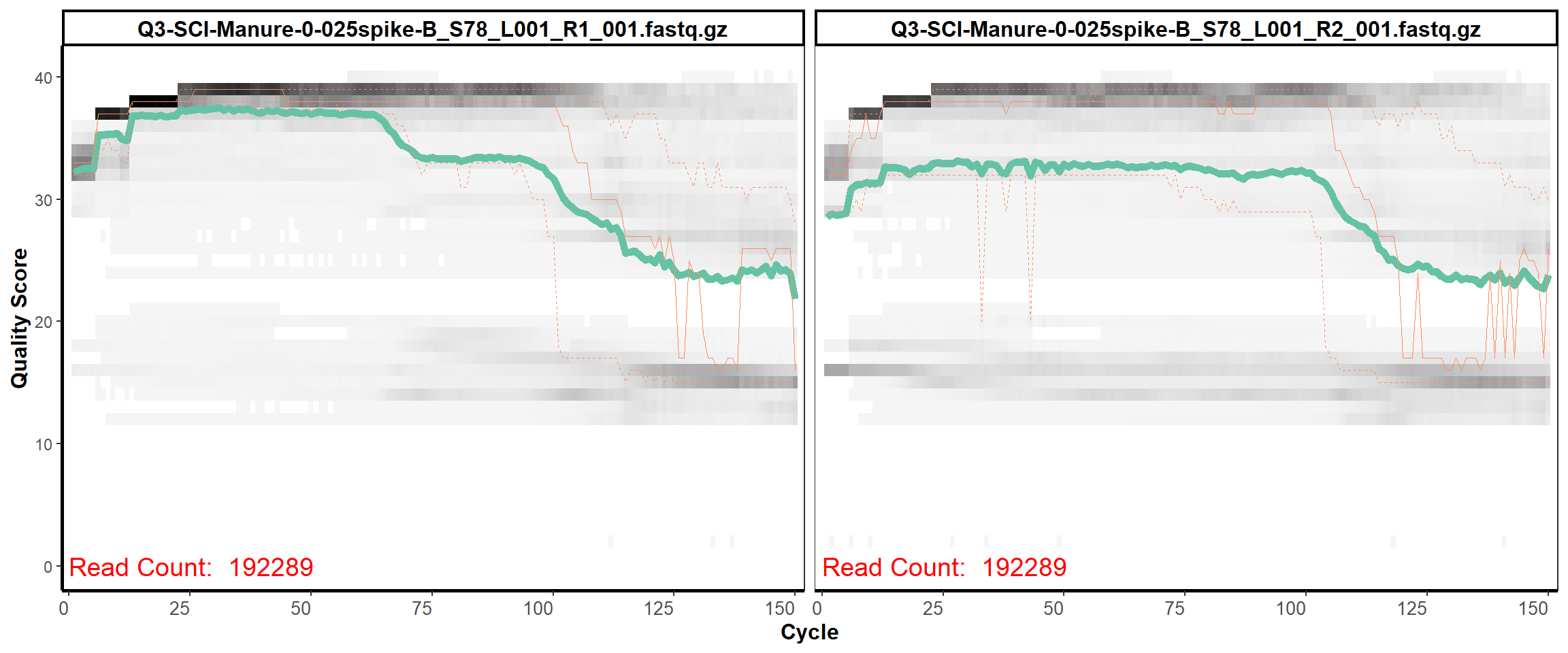
3
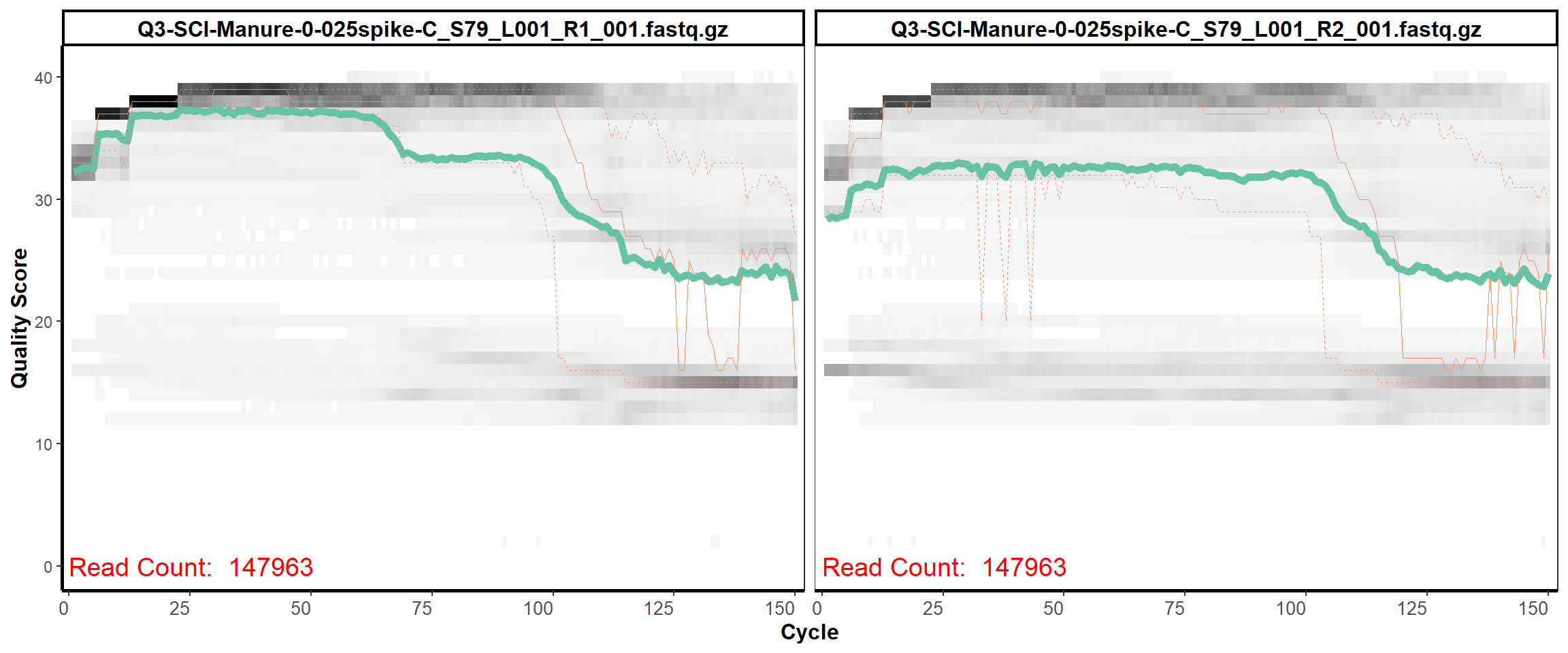
4

5
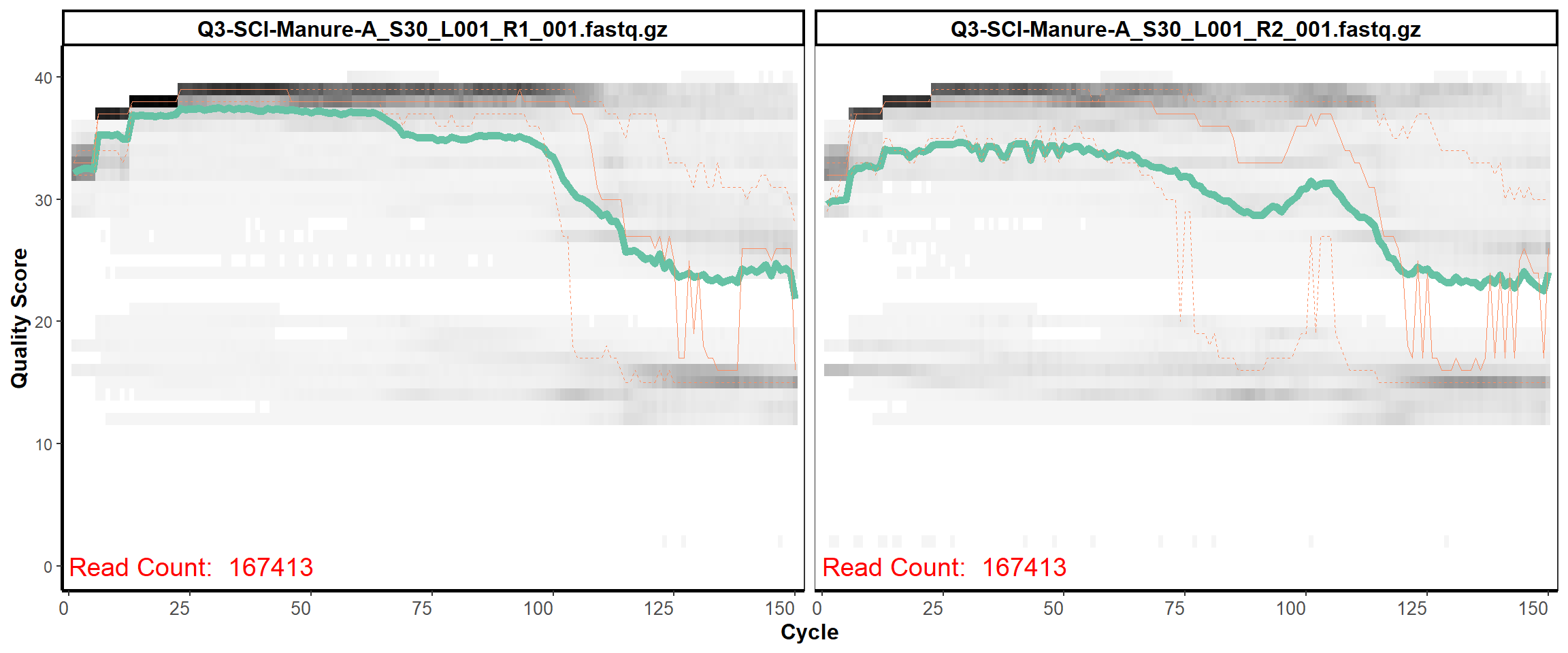
6

7
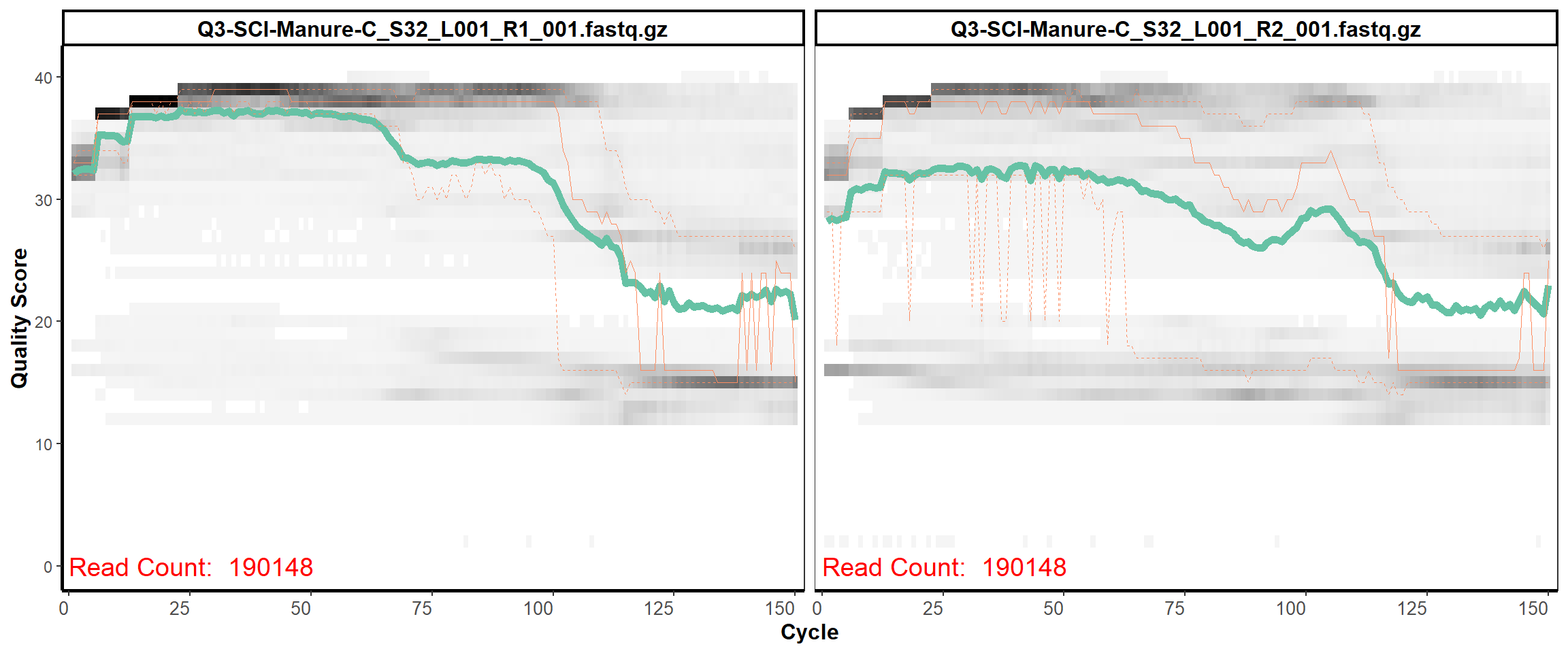
8
9

10
11
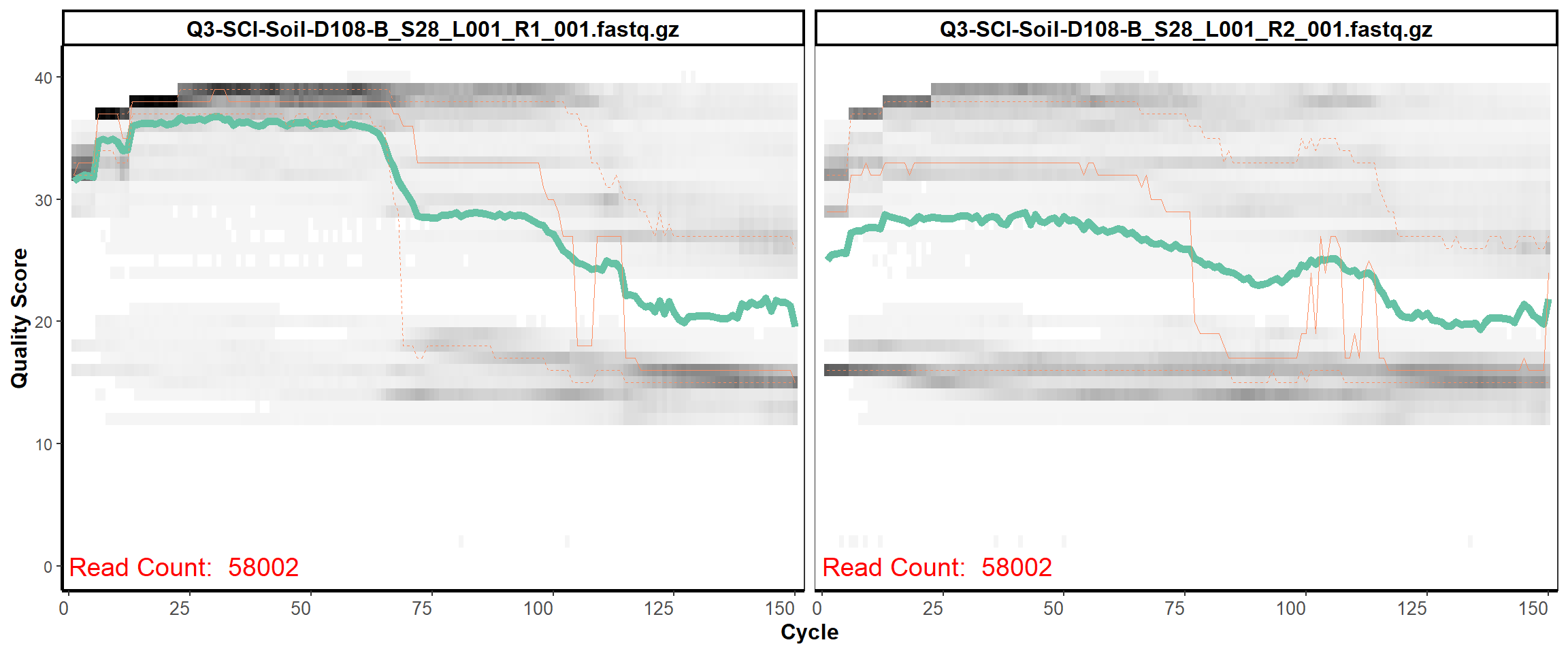
12
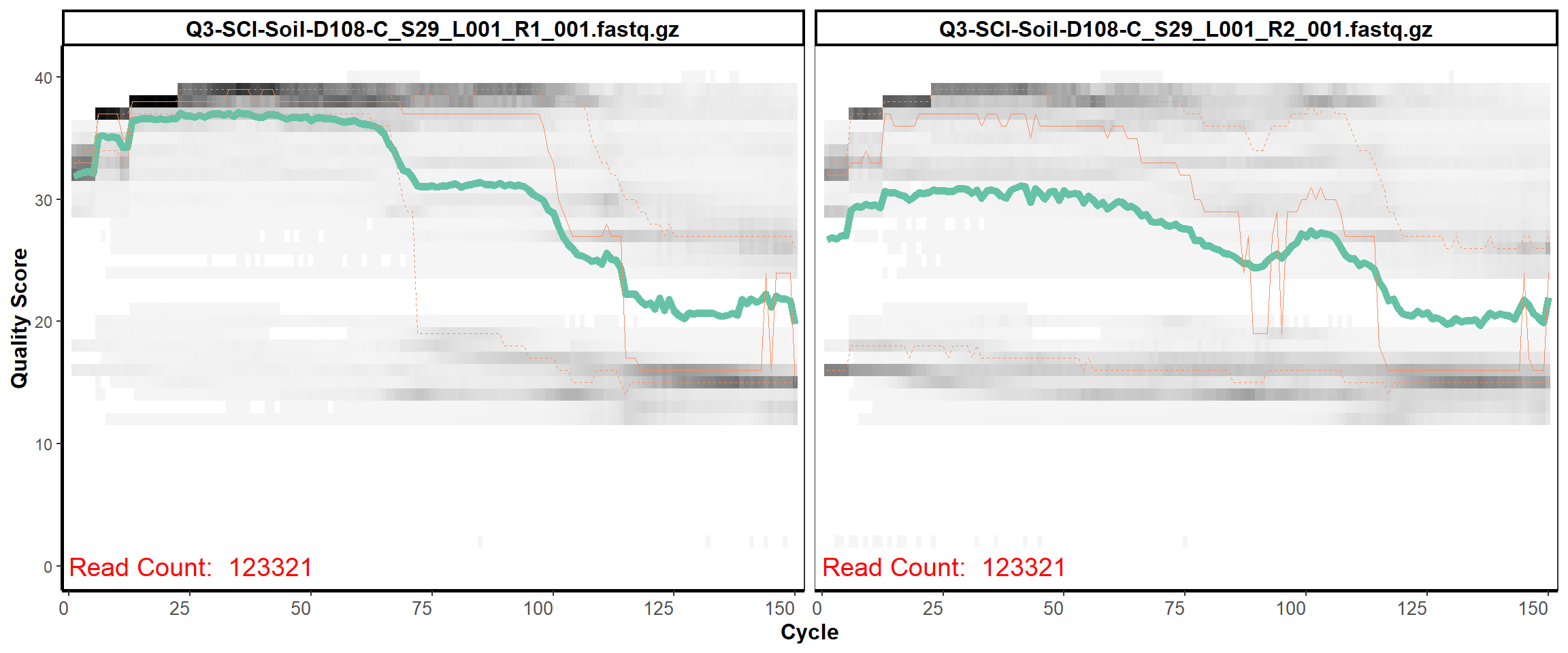
13
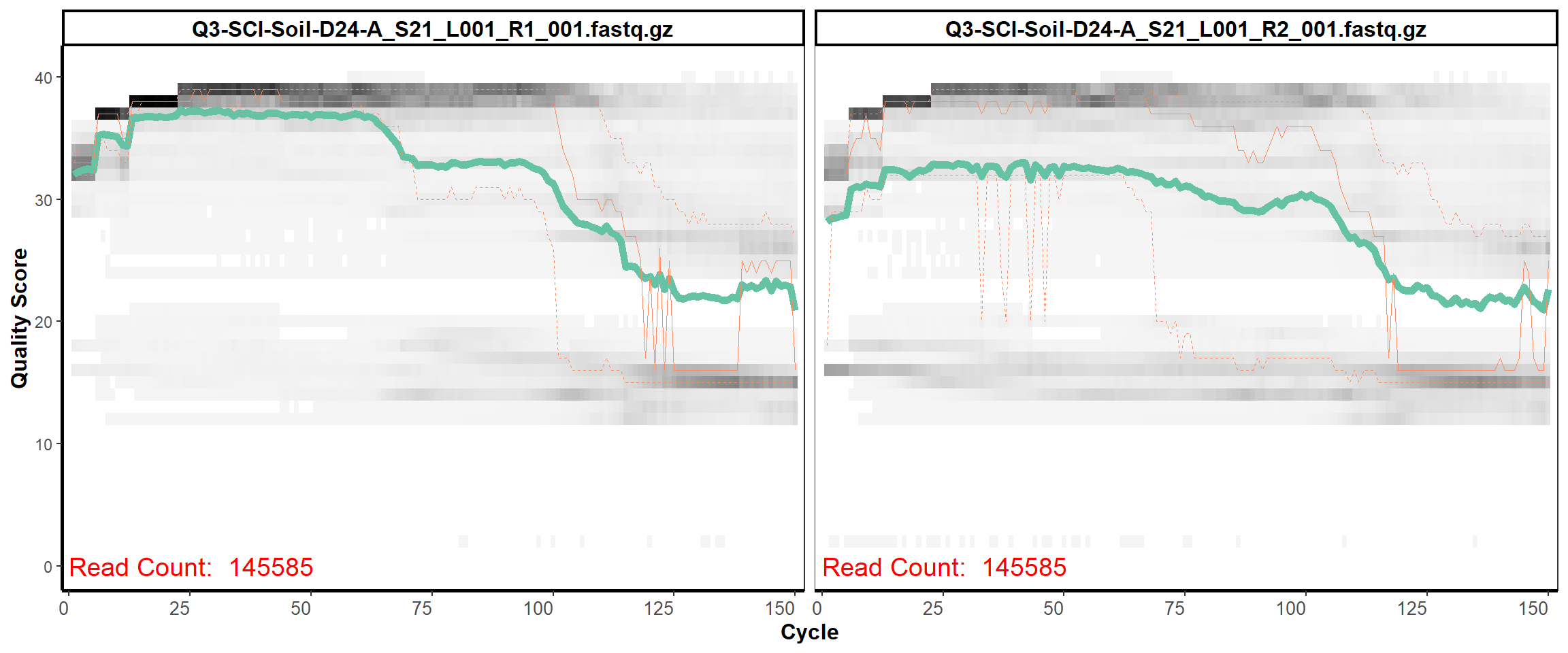
14
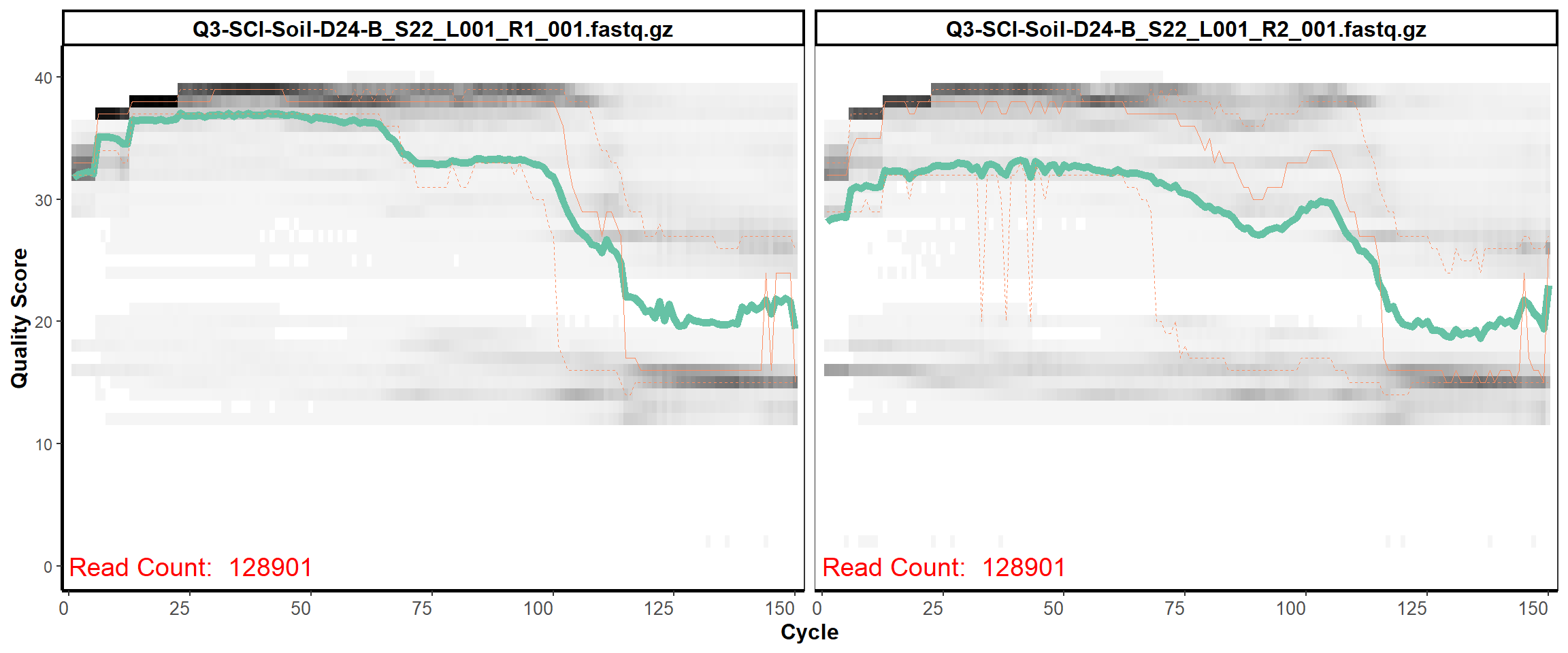
15
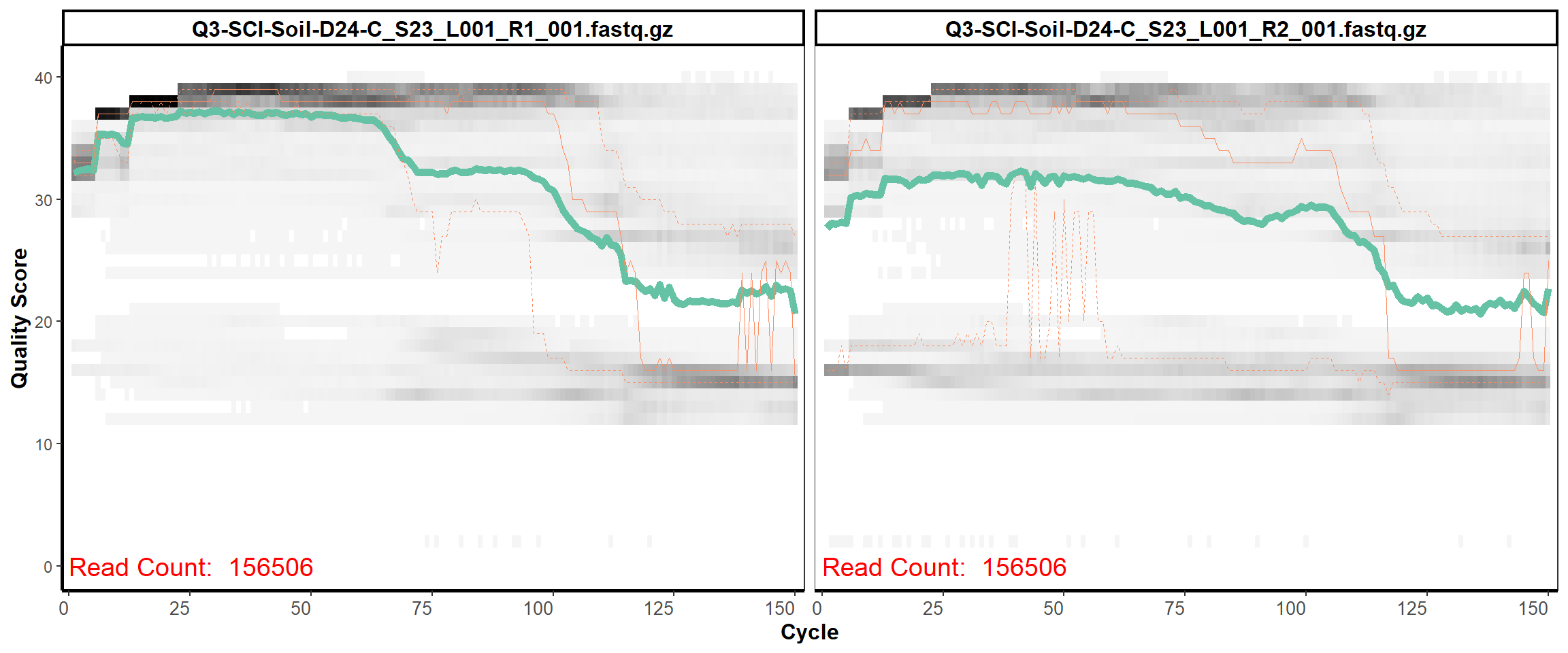
16
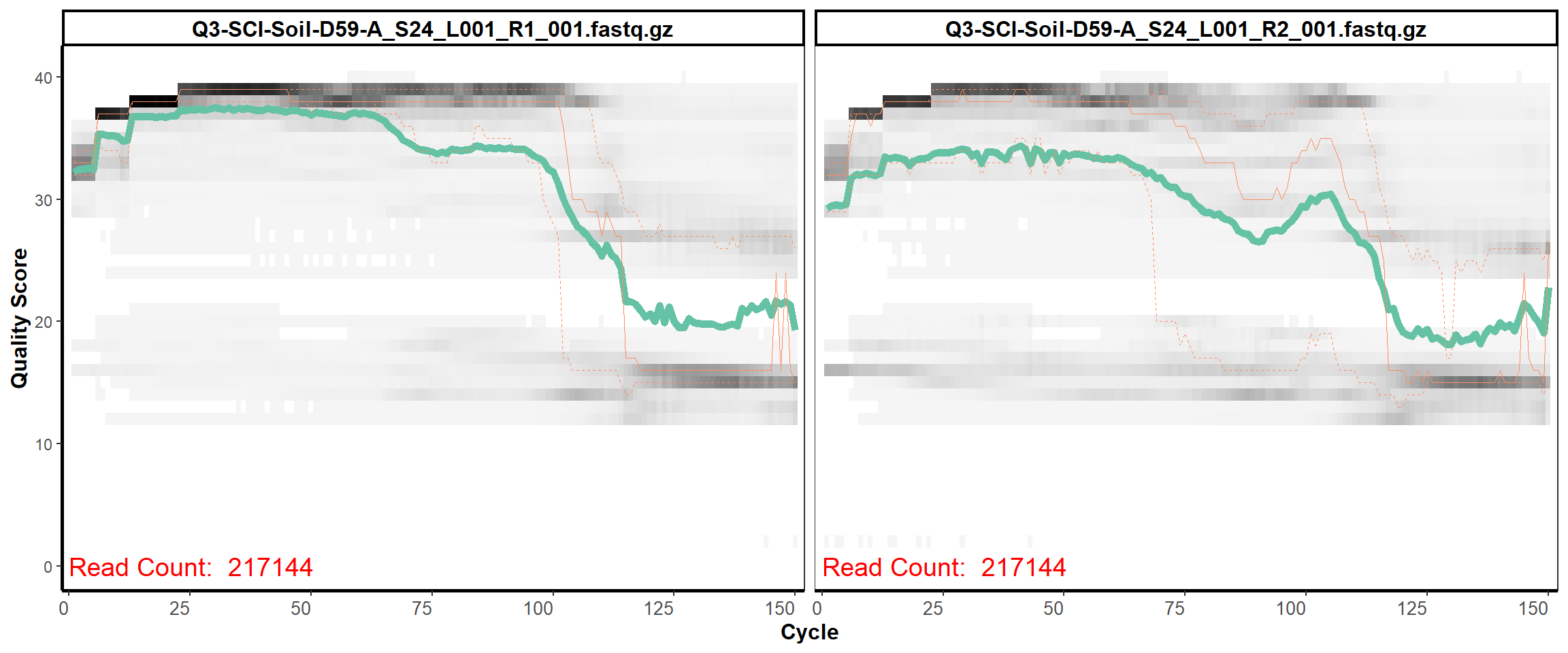
17
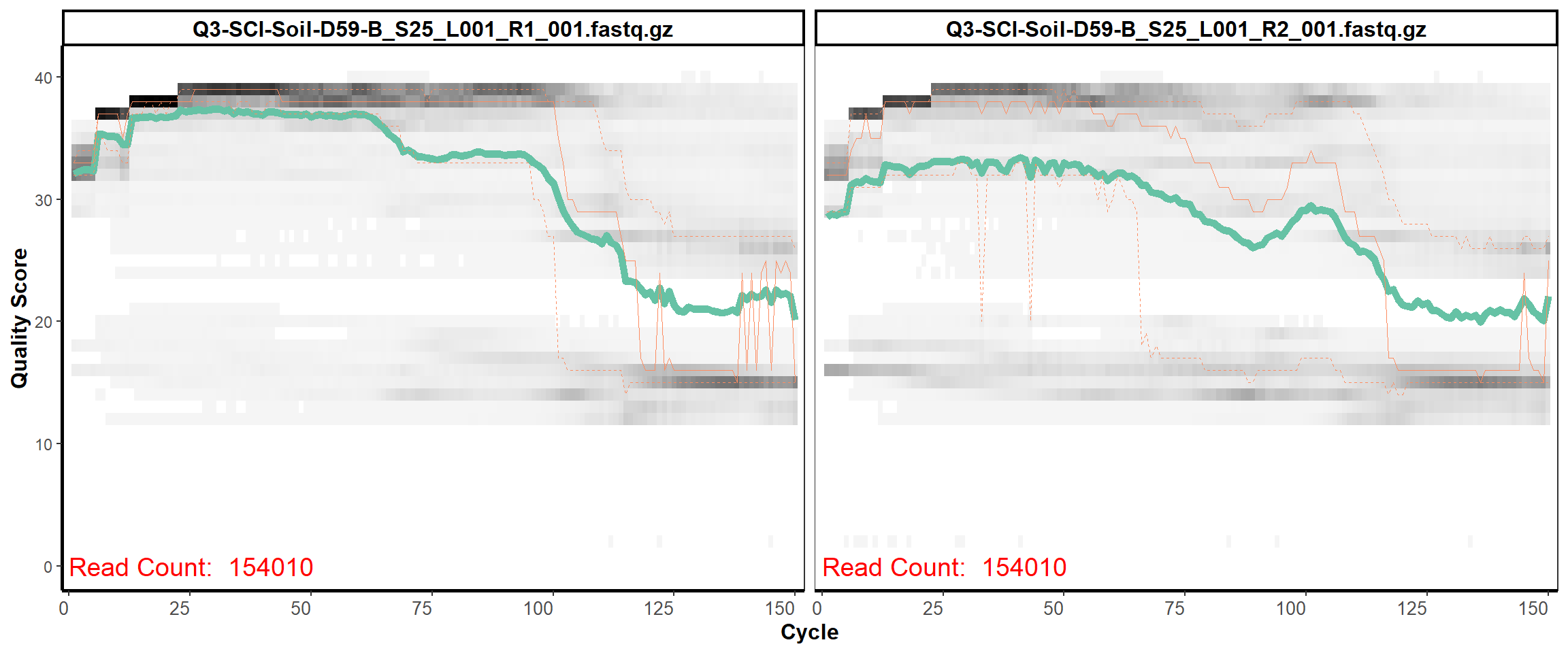
18

Sequence Results
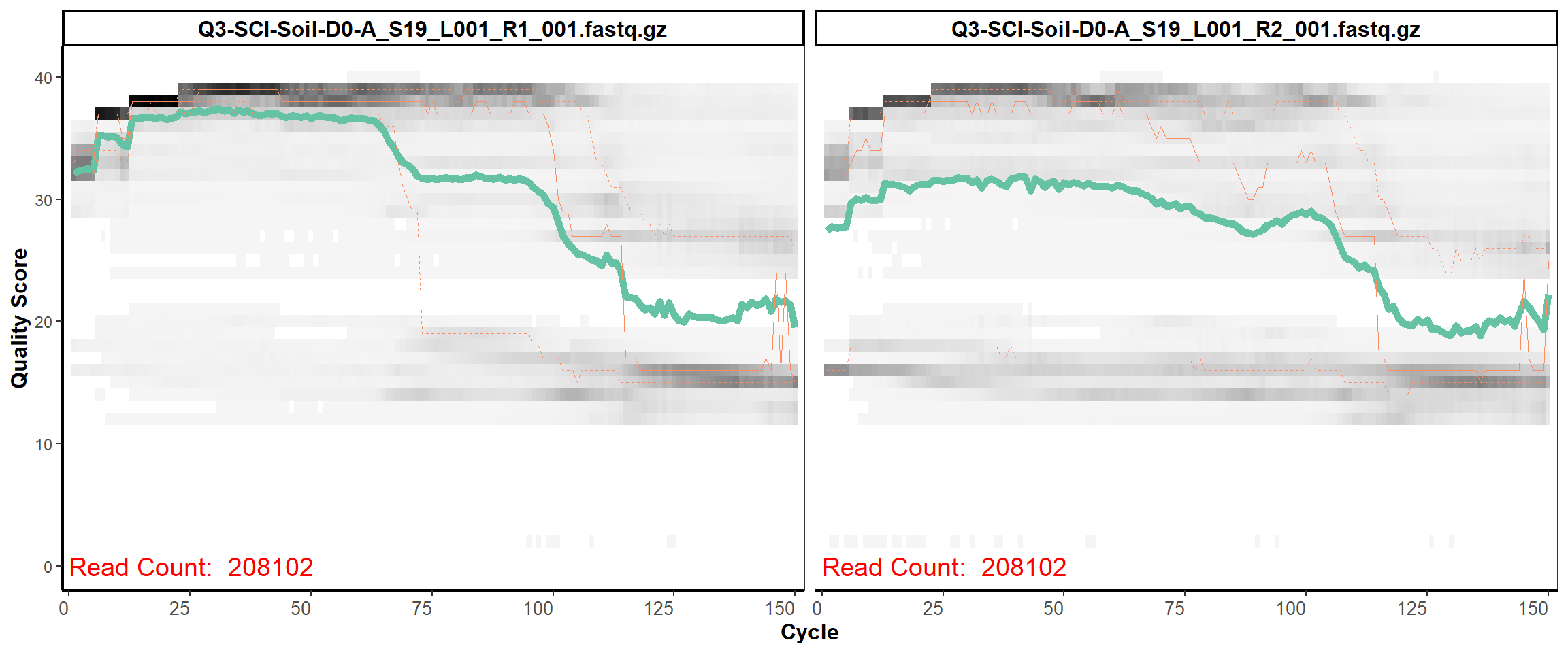
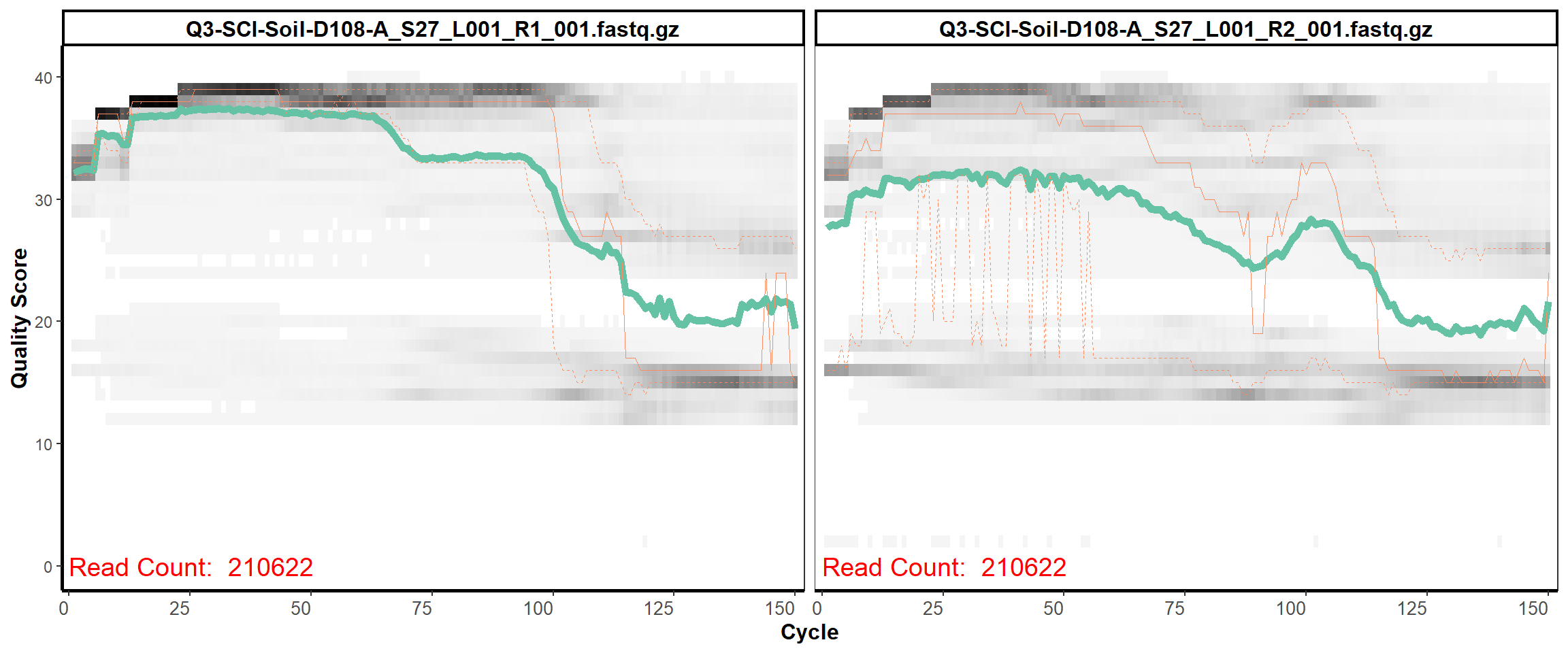
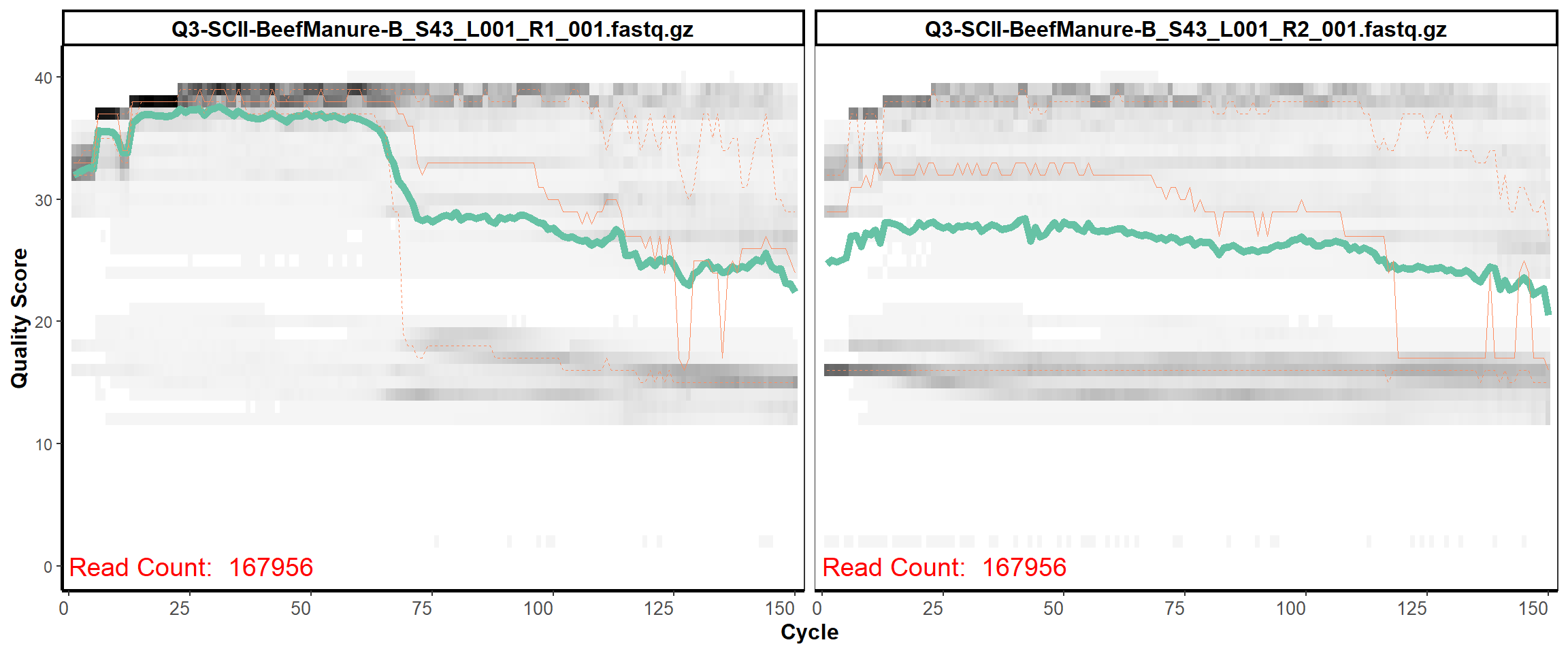
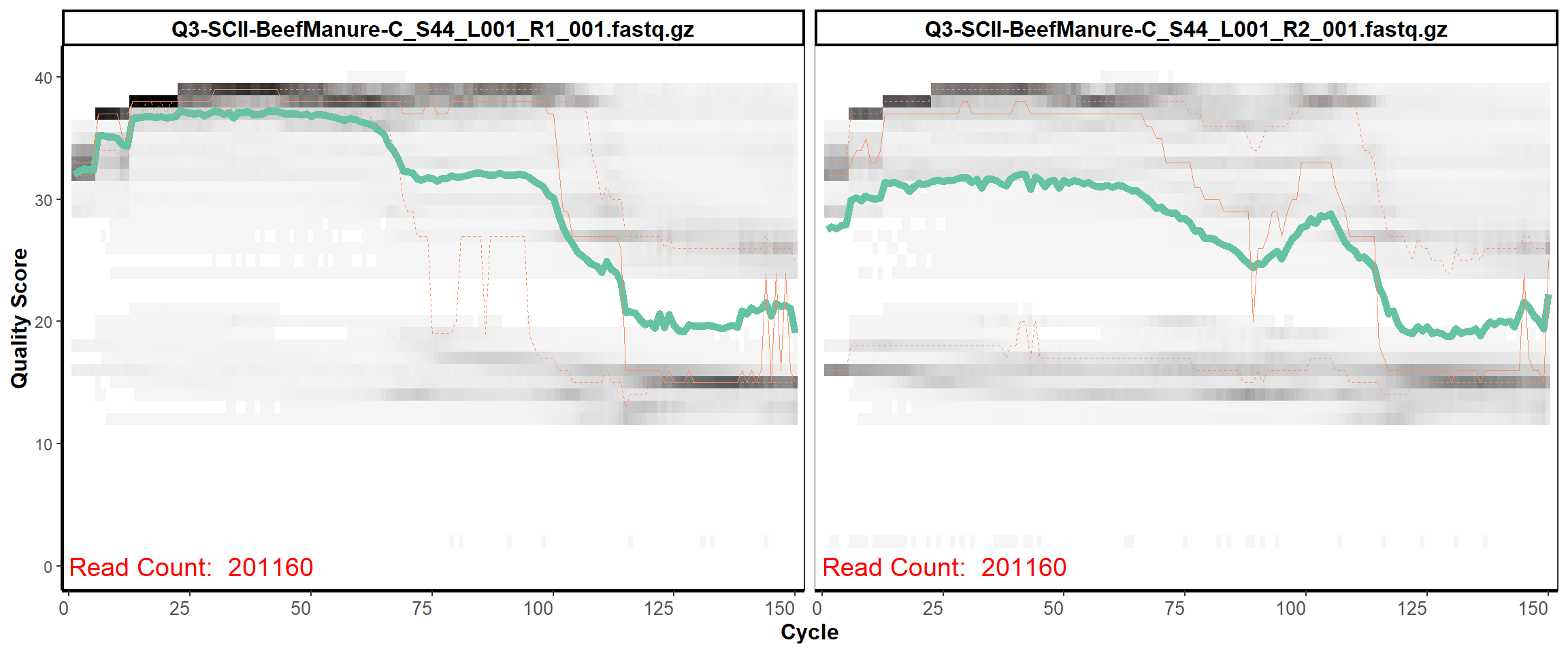
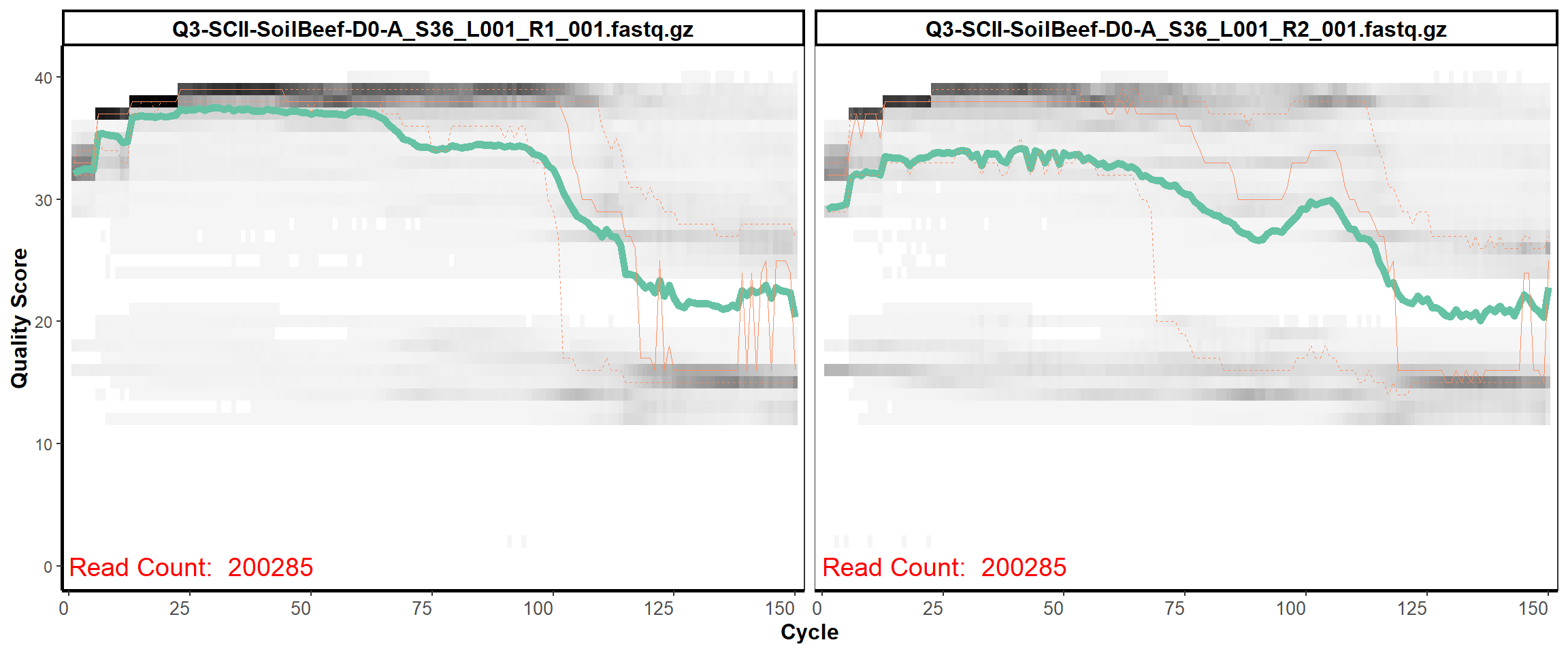
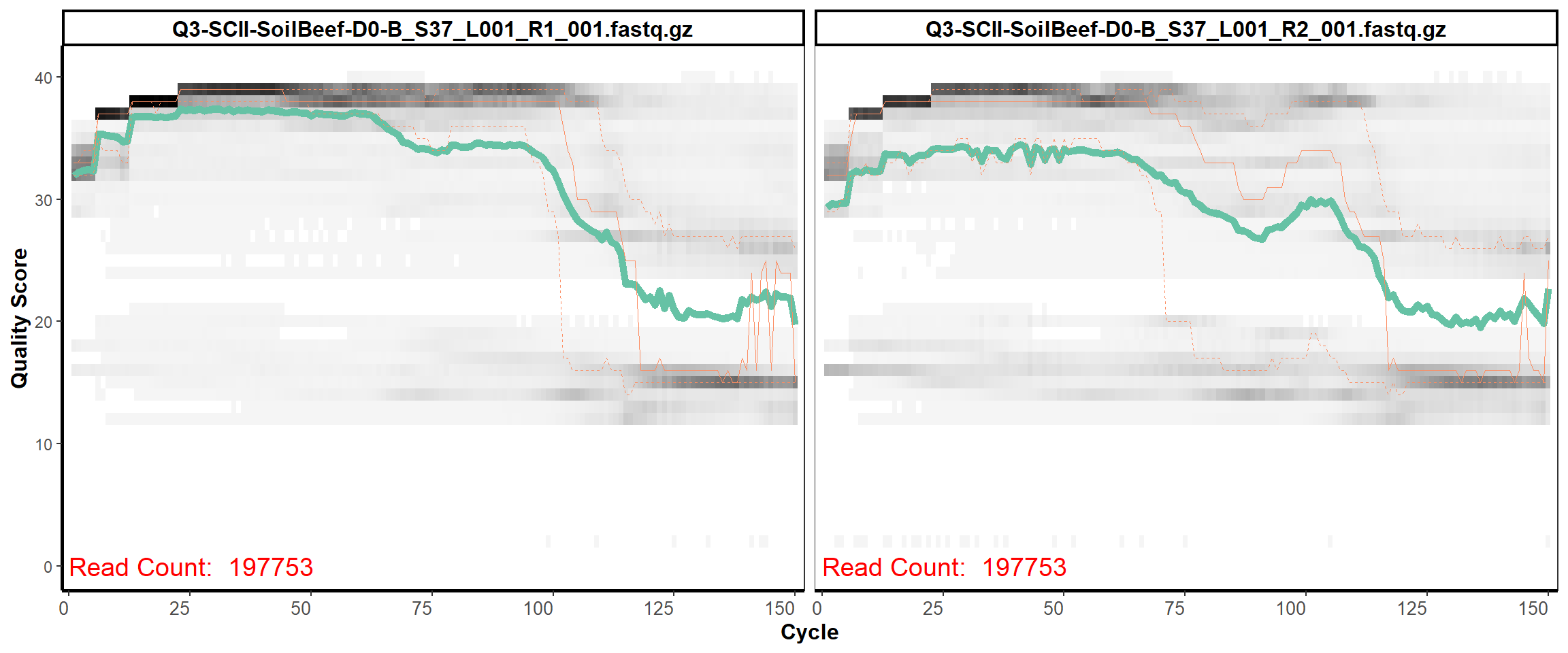
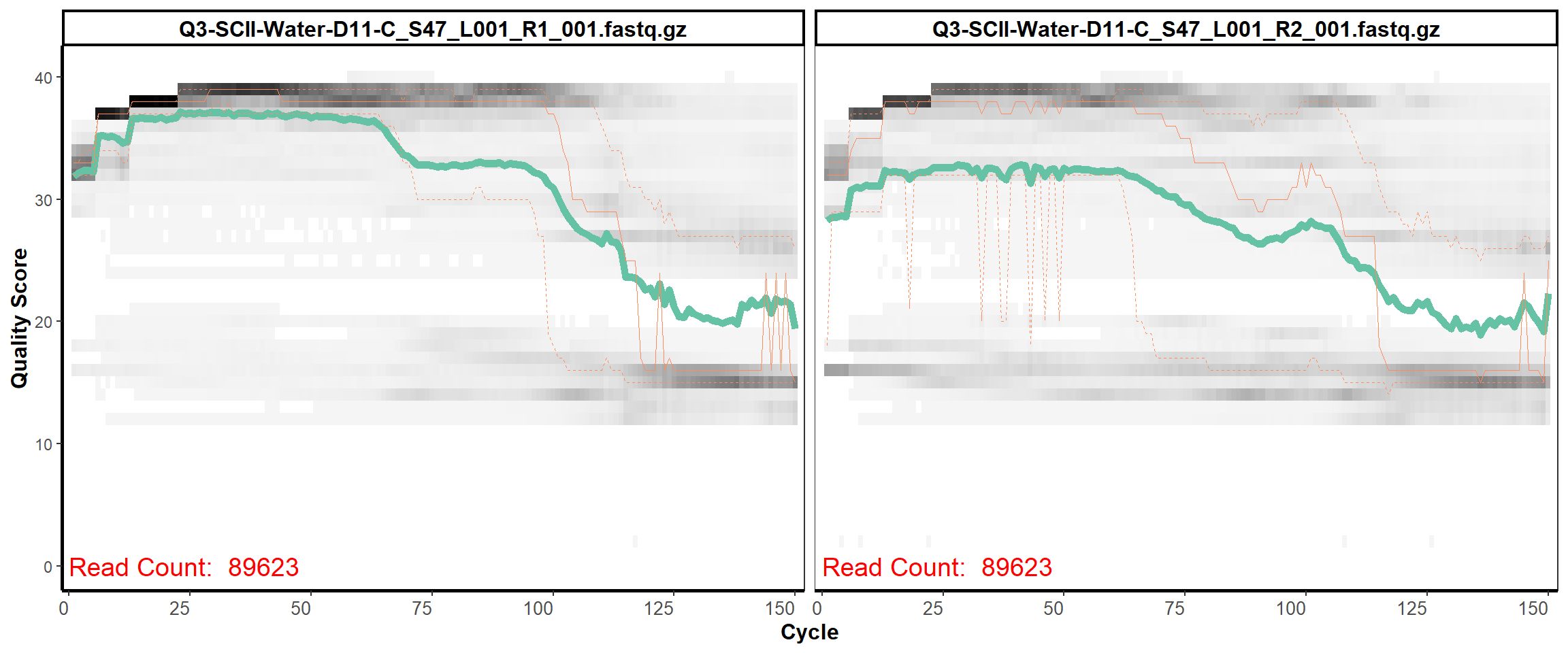
Soil Column II
MiSeq Quality scores
1
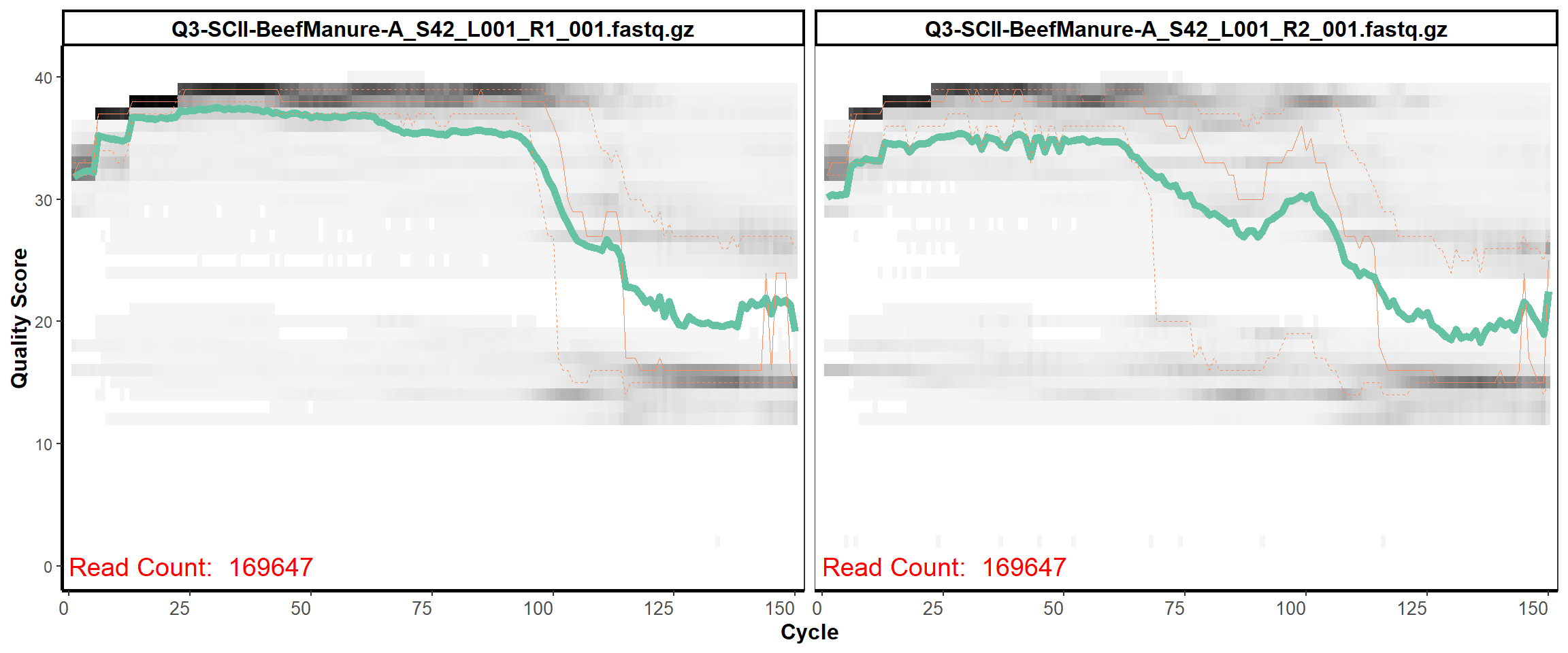
2
3
4
5
6

7

8
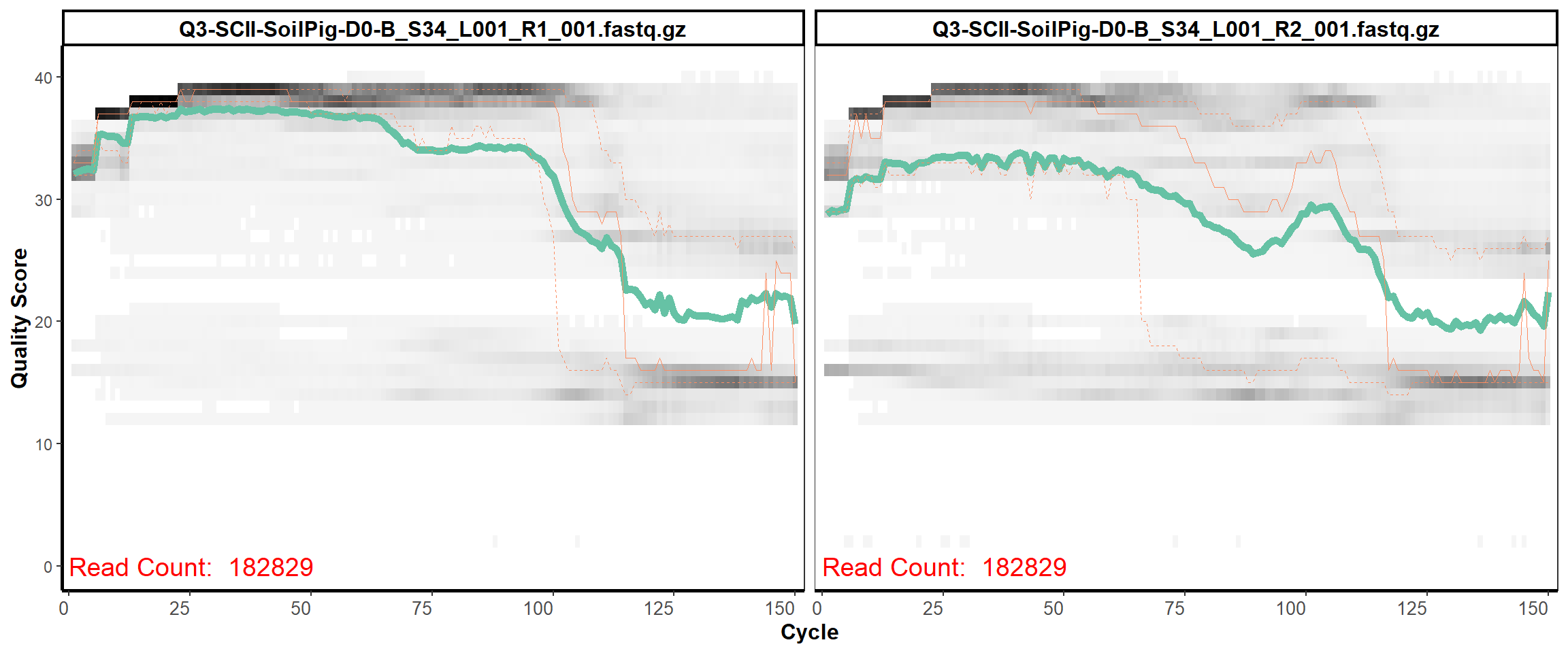
9

10
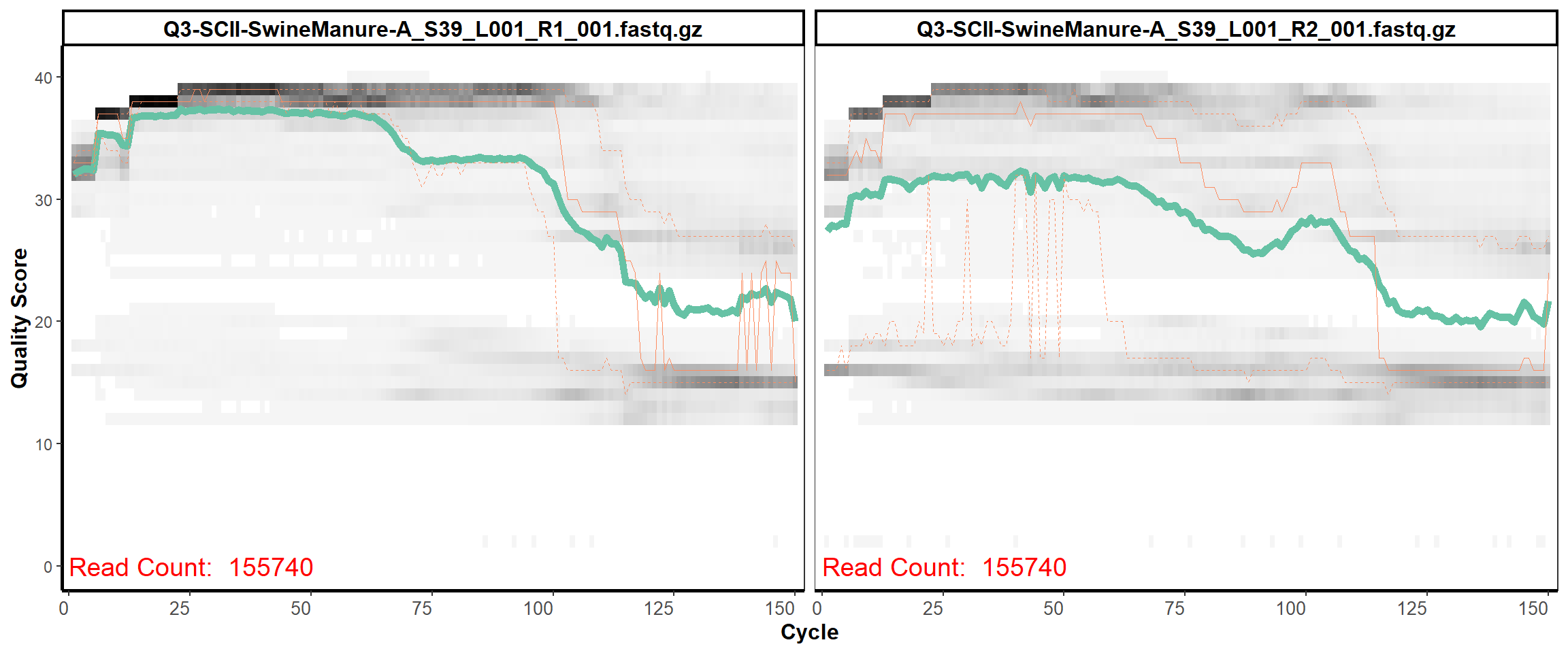
11
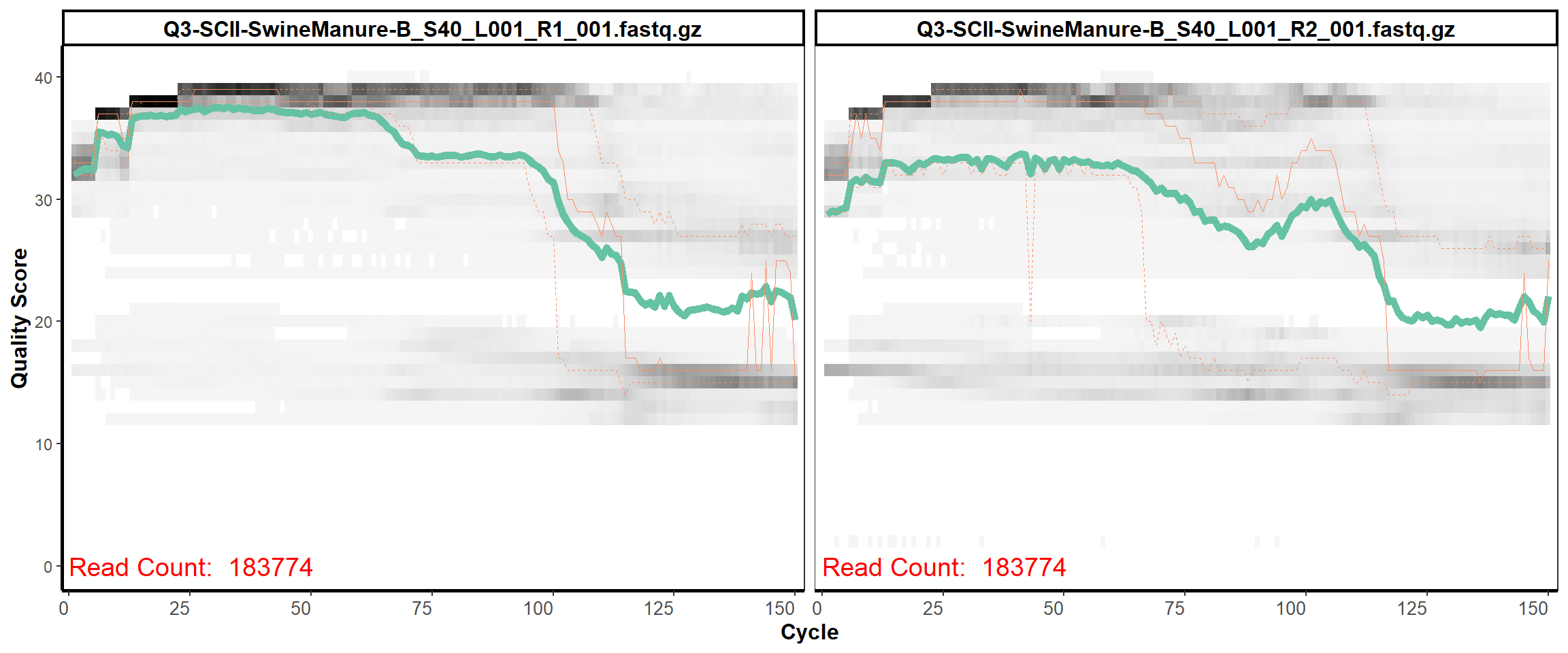
12
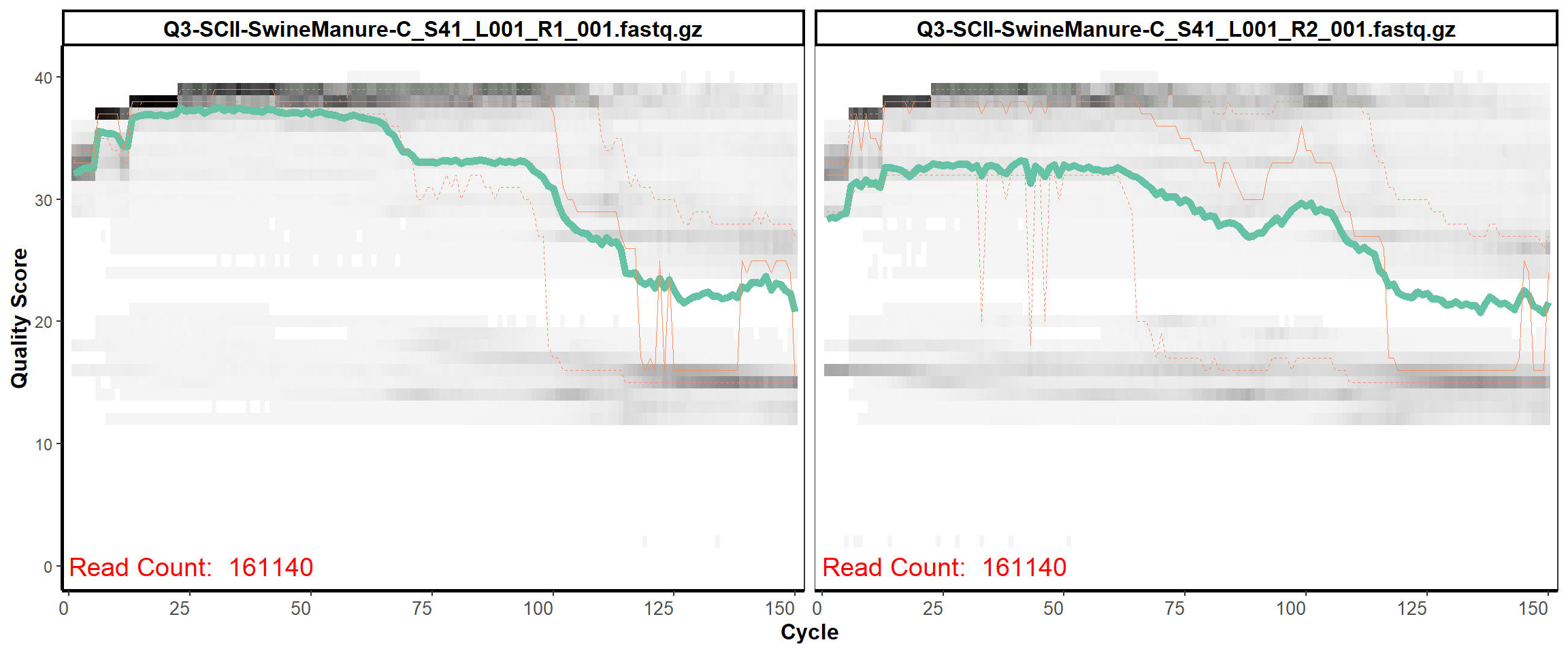
13
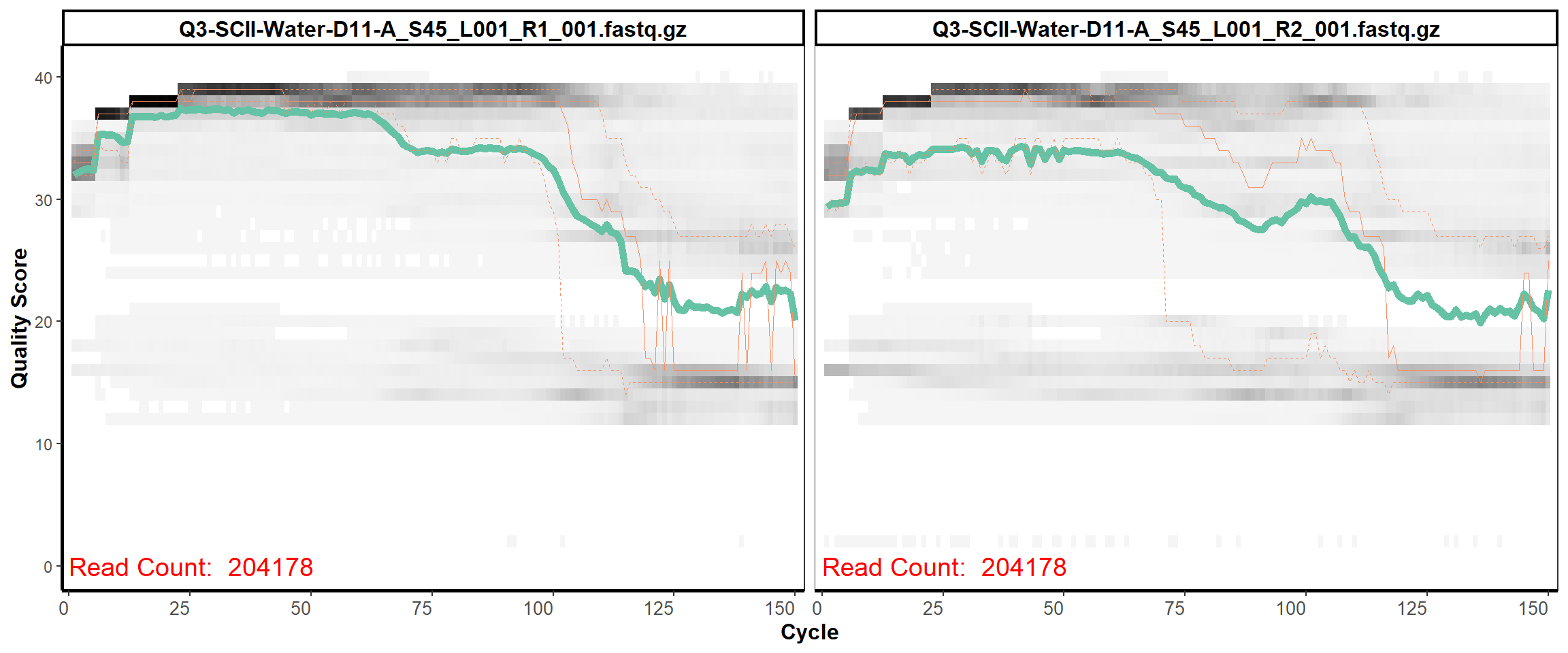
14

15
Sequence Results
Swine Fecal
MiSeq Quality scores
1
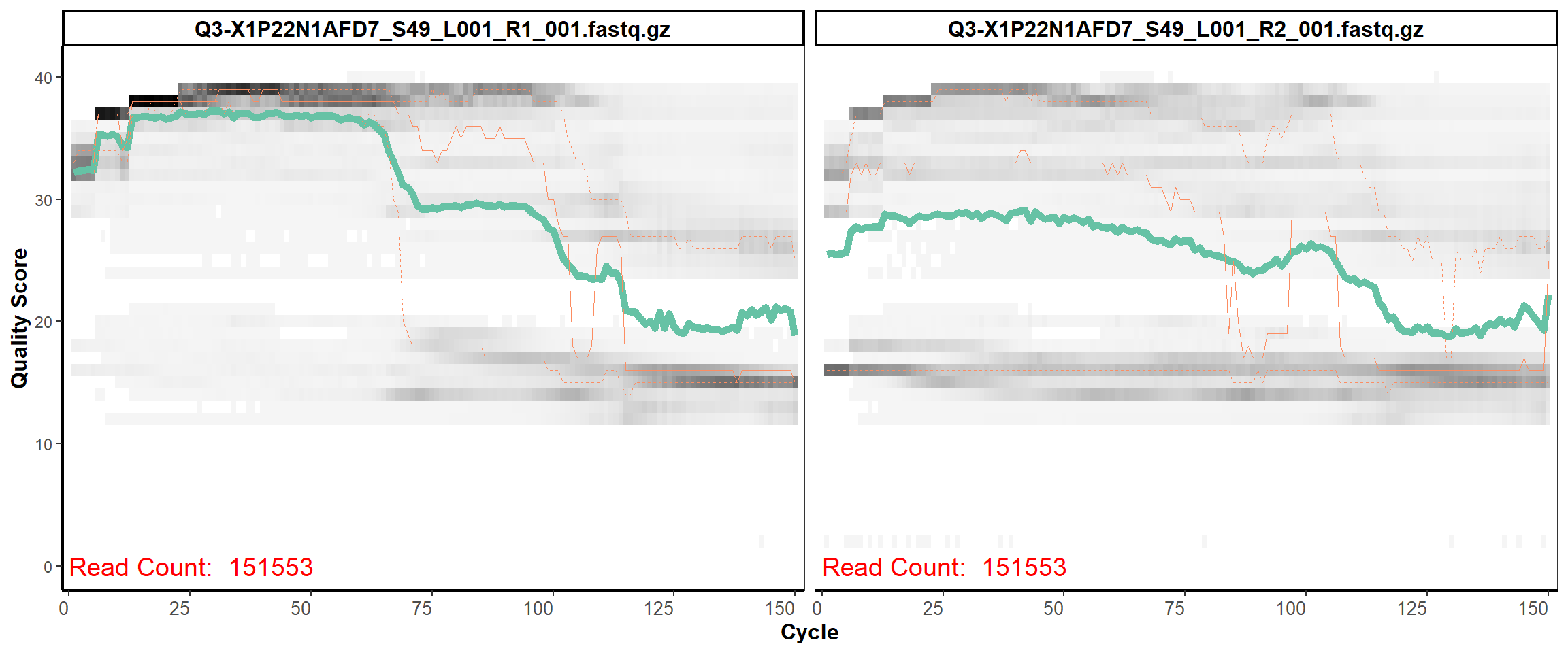
2
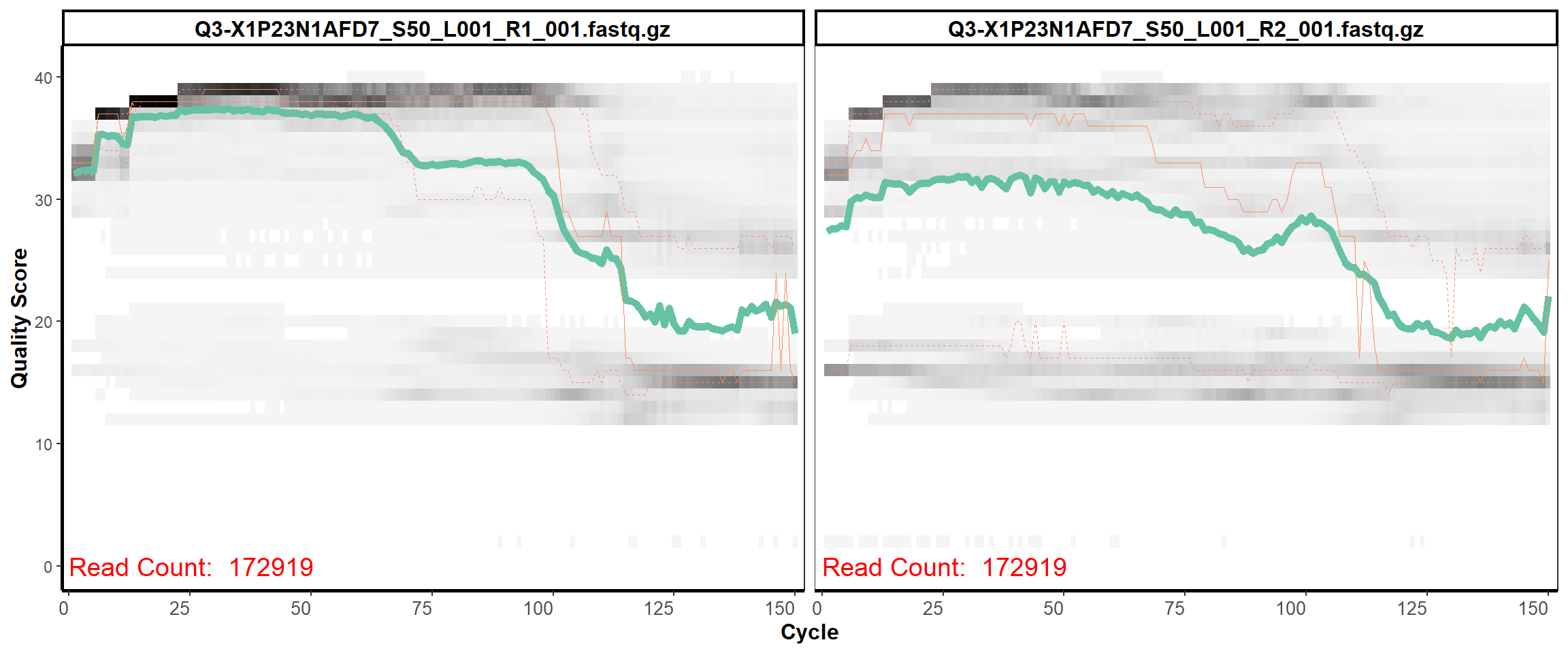
3

4
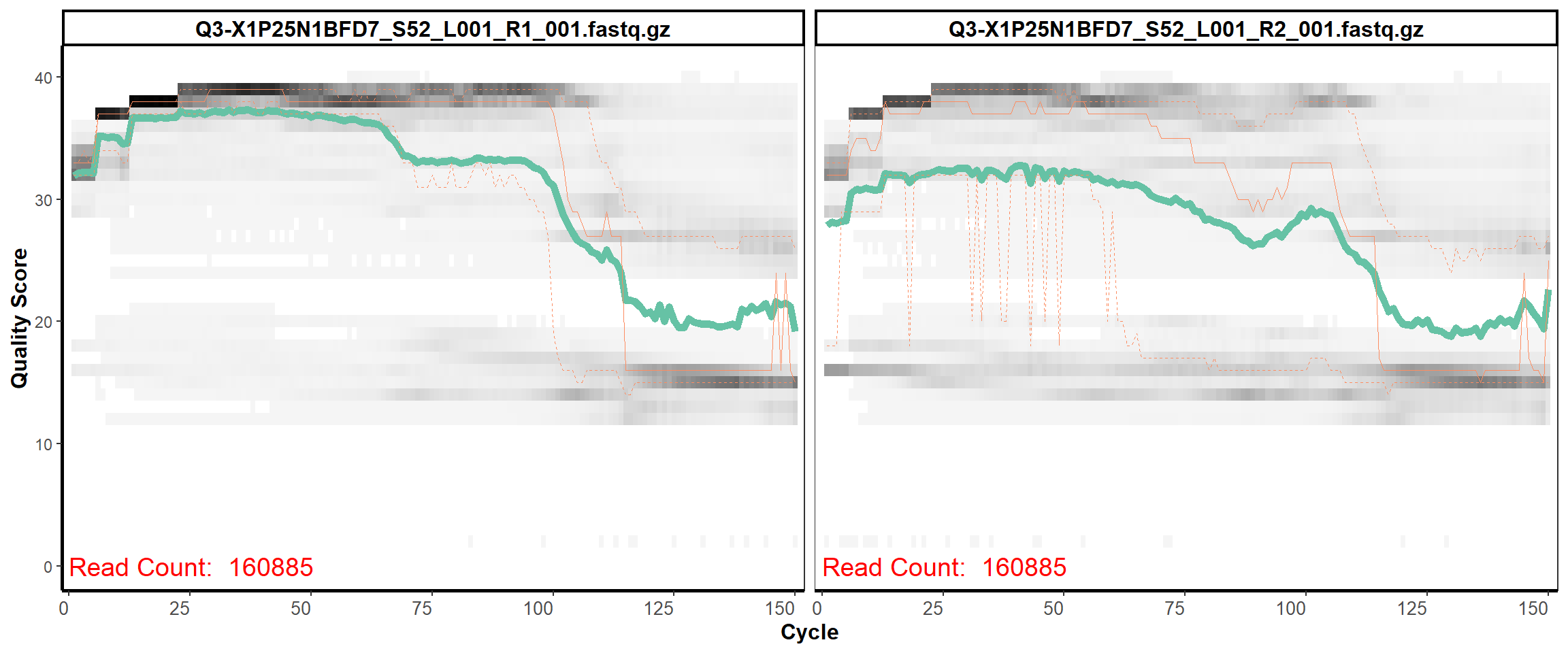
5
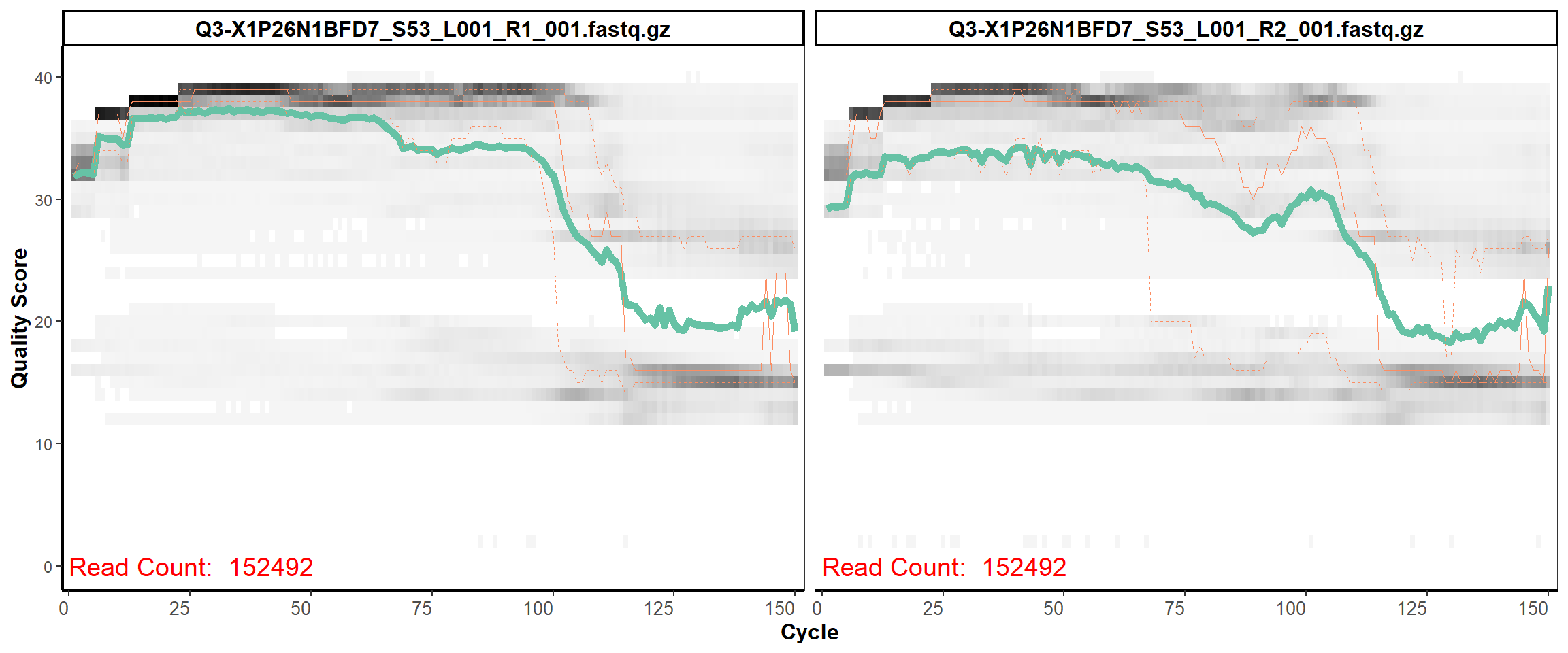
6

7
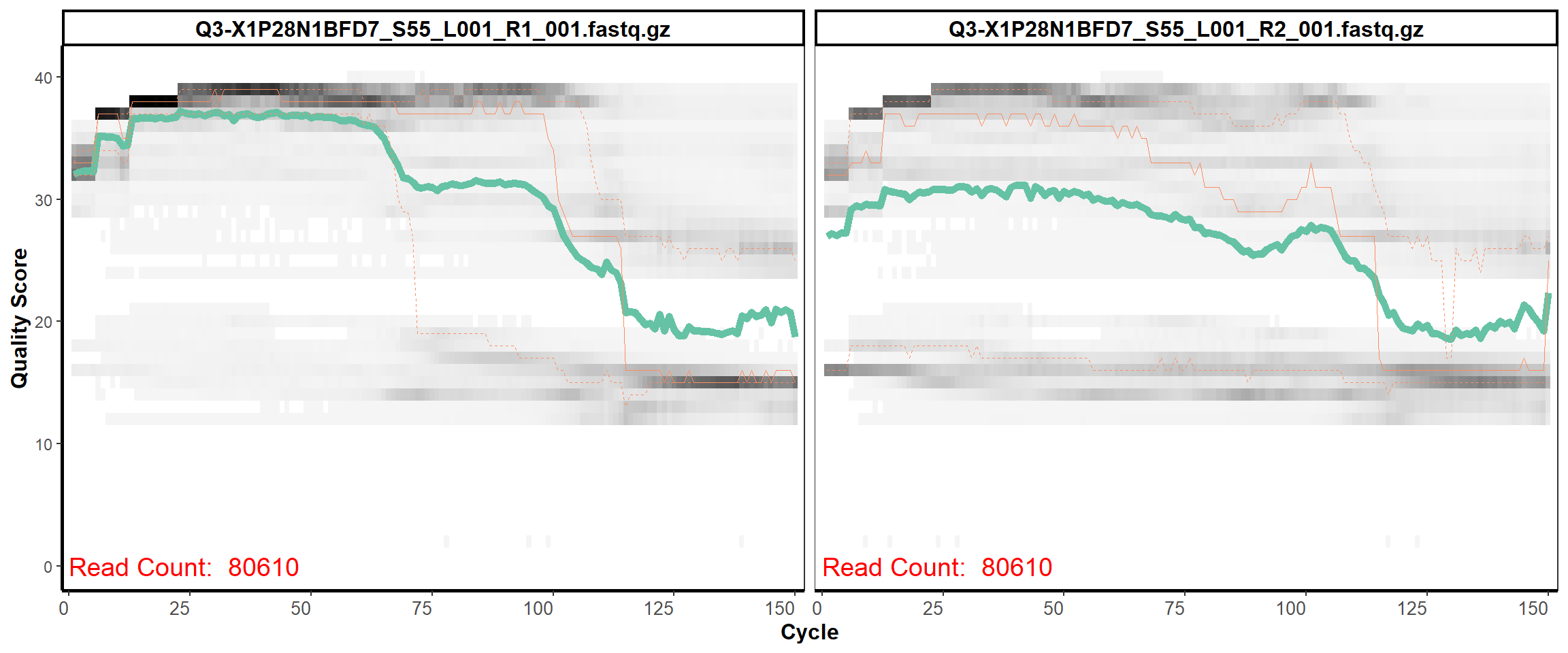
8
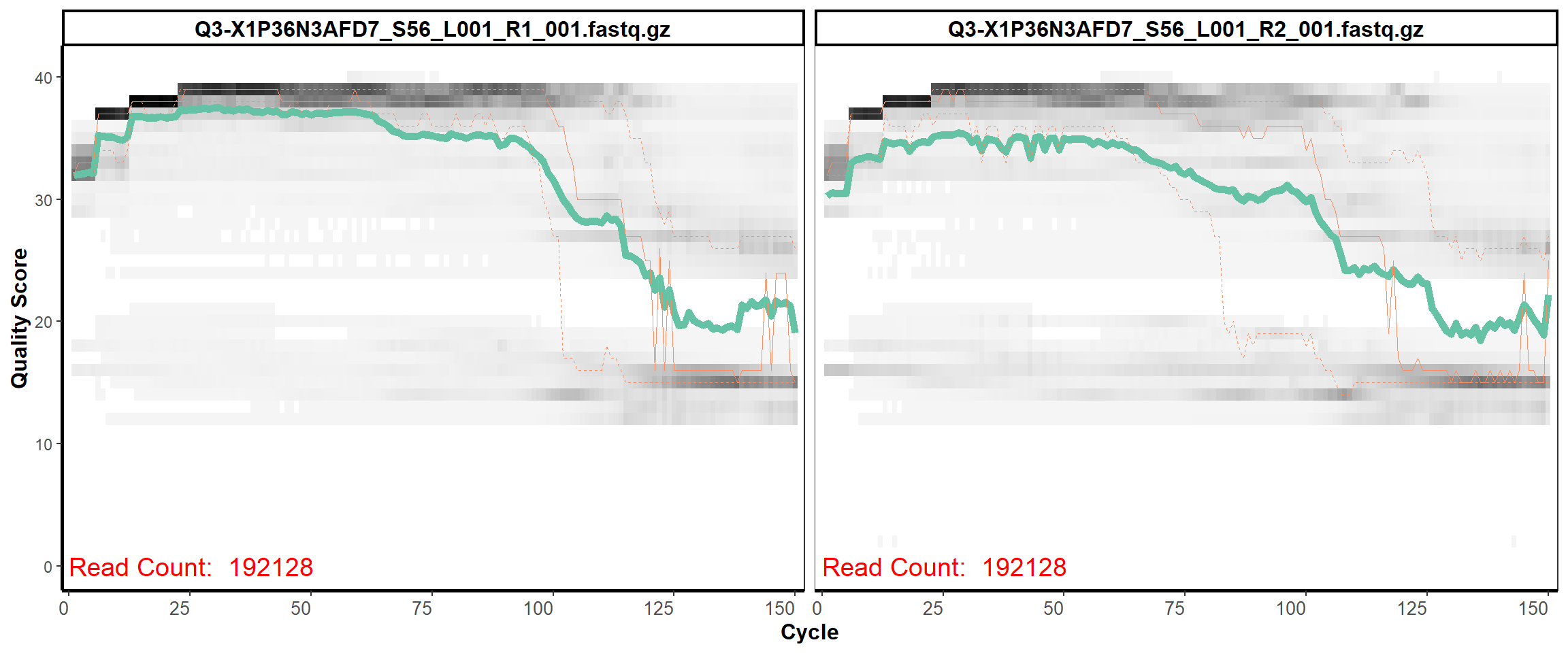
9
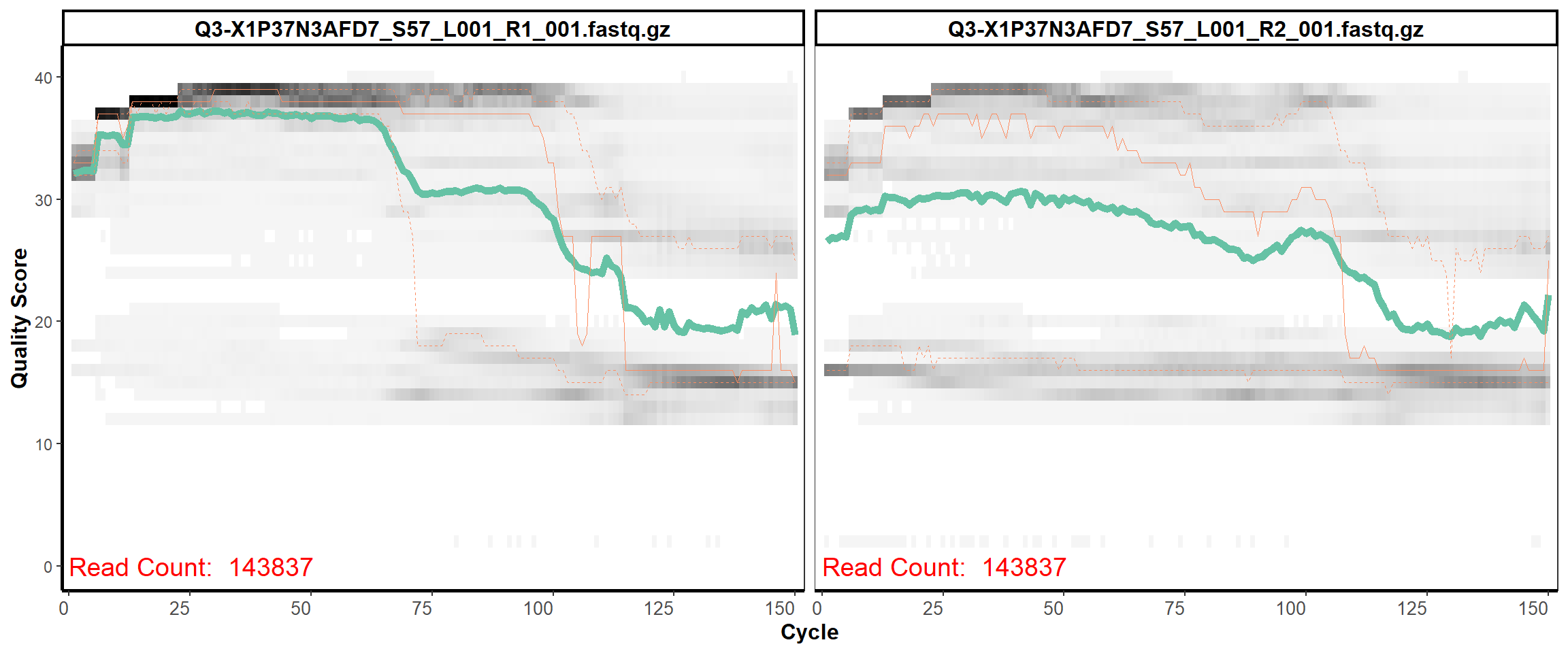
10

11
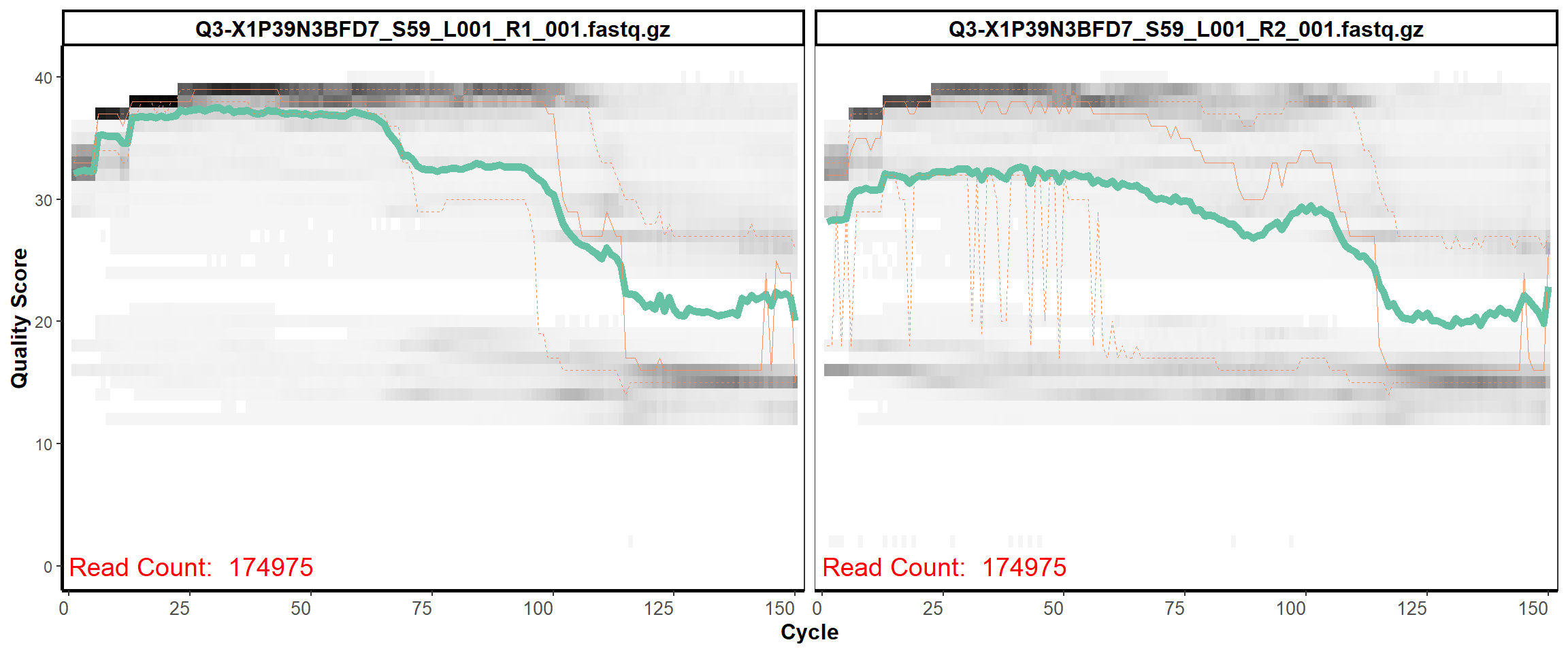
12
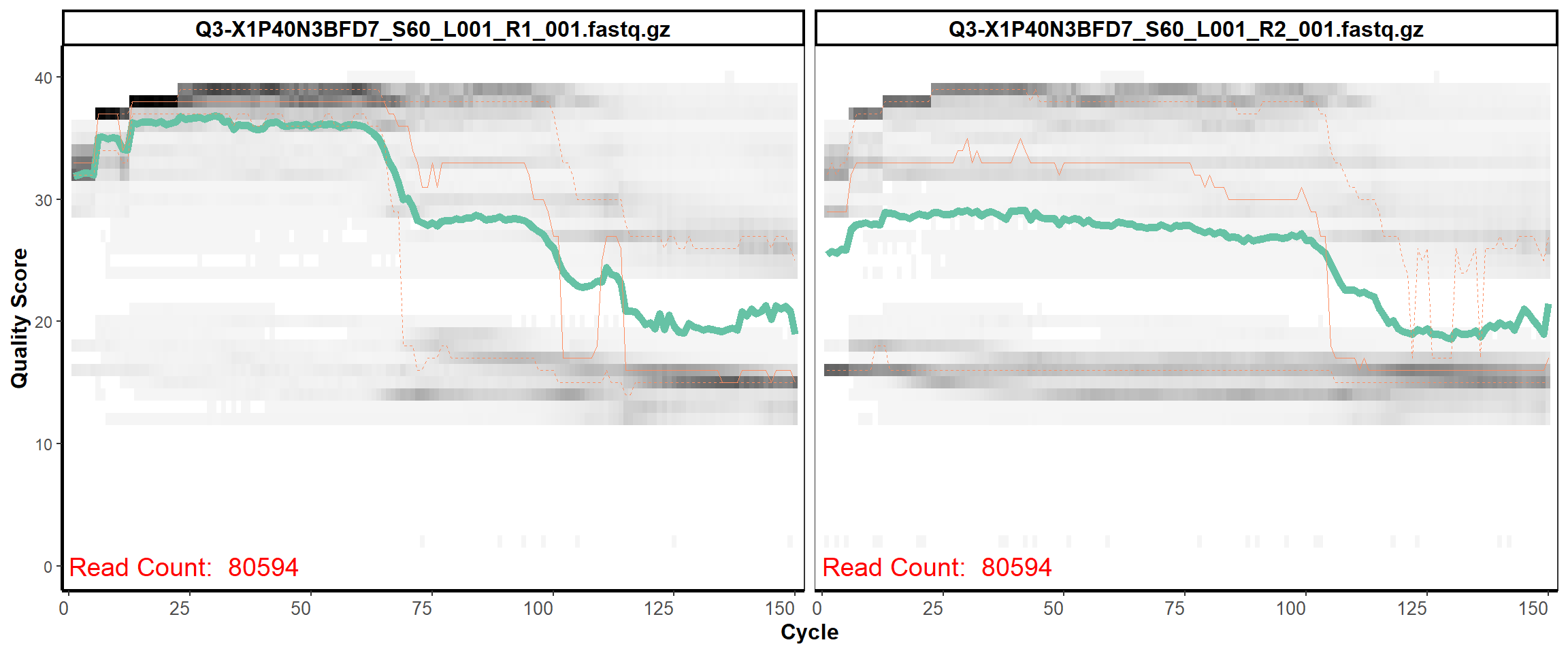
13
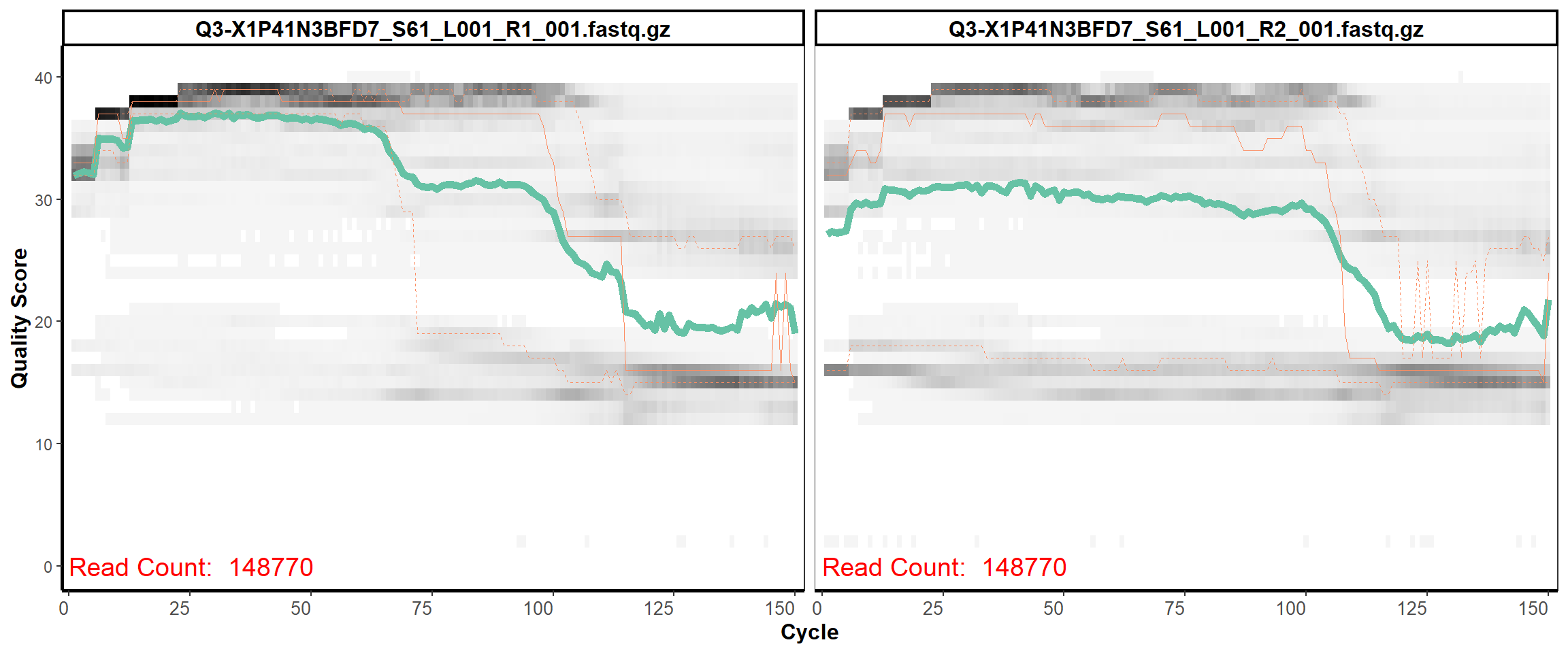
14
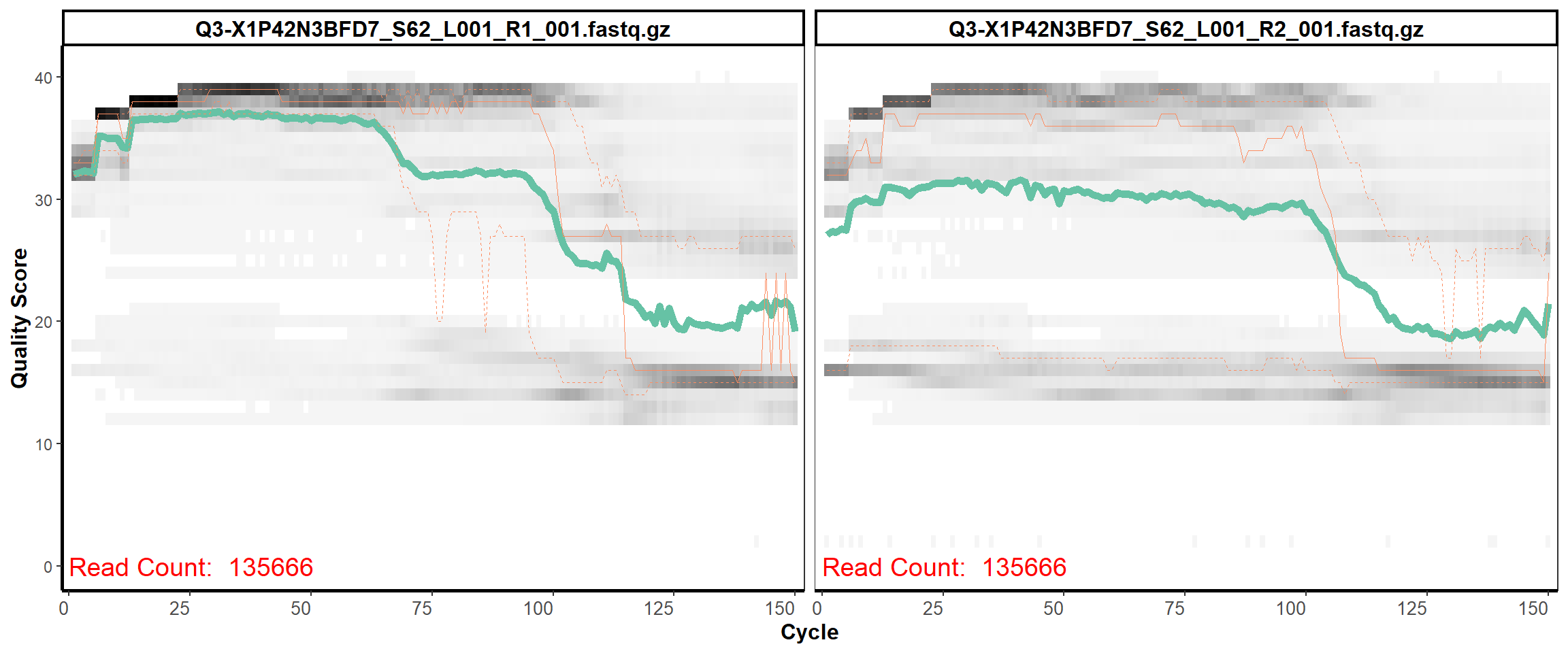
15
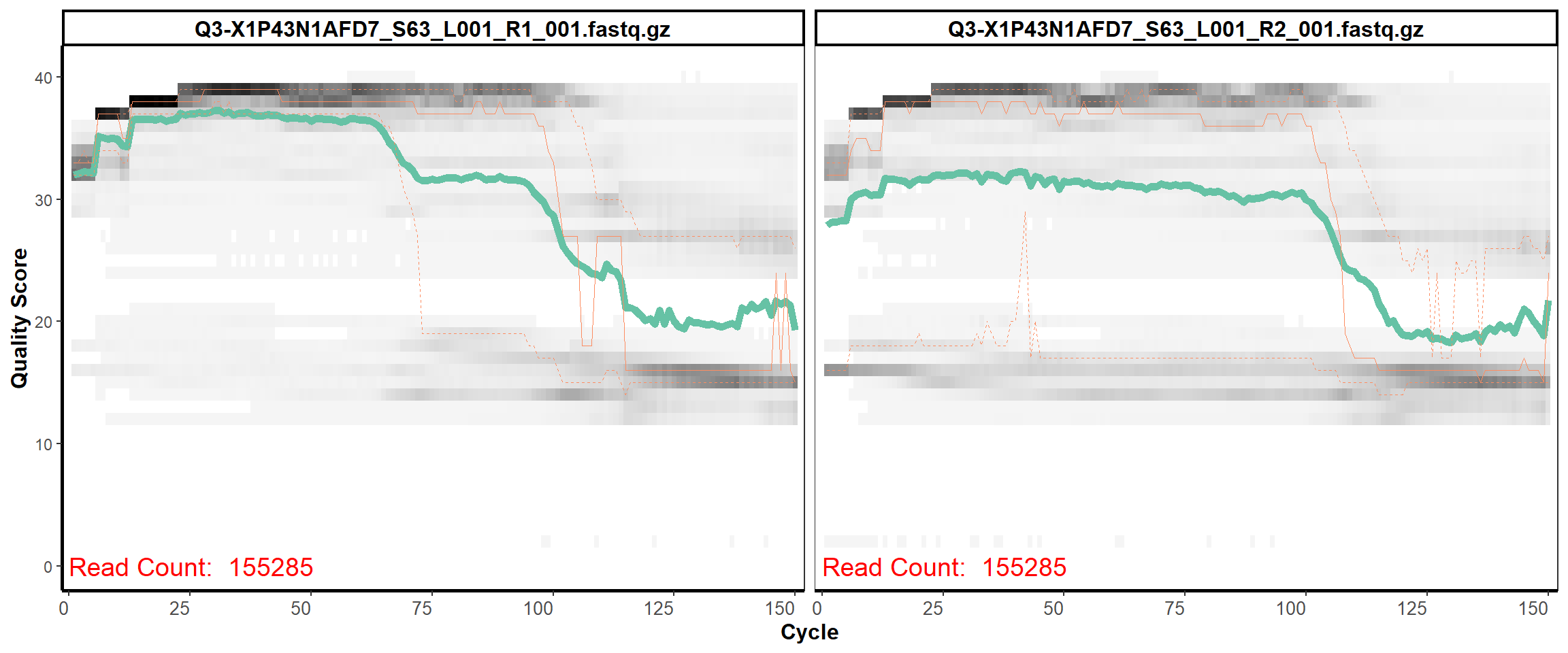
16
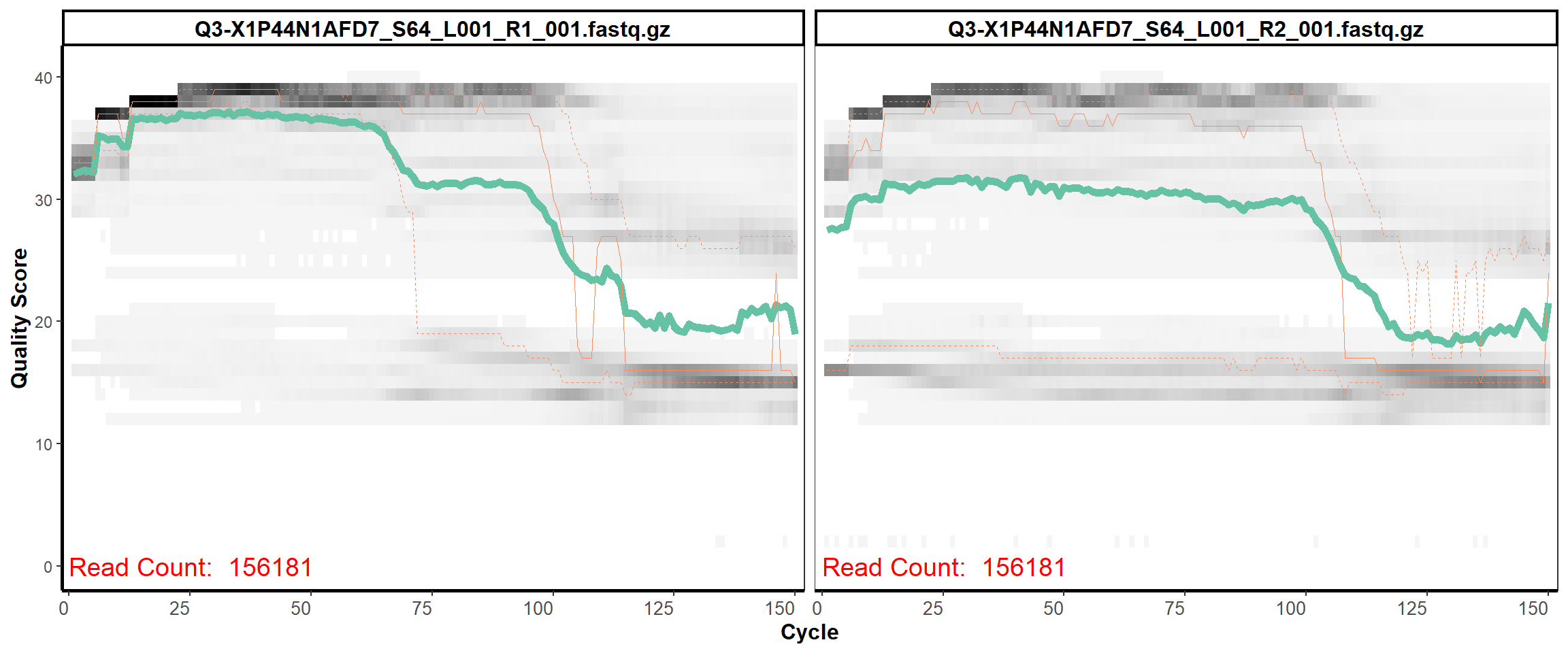
17
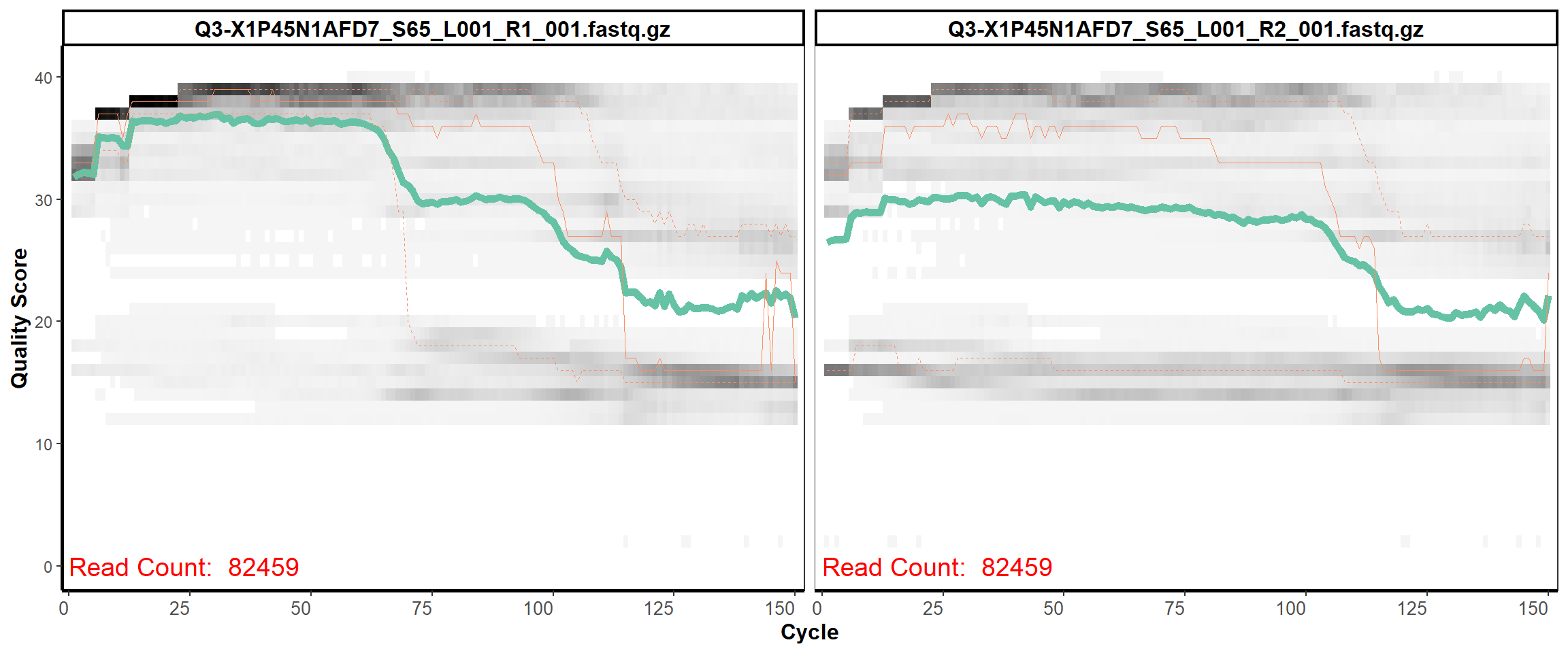
18
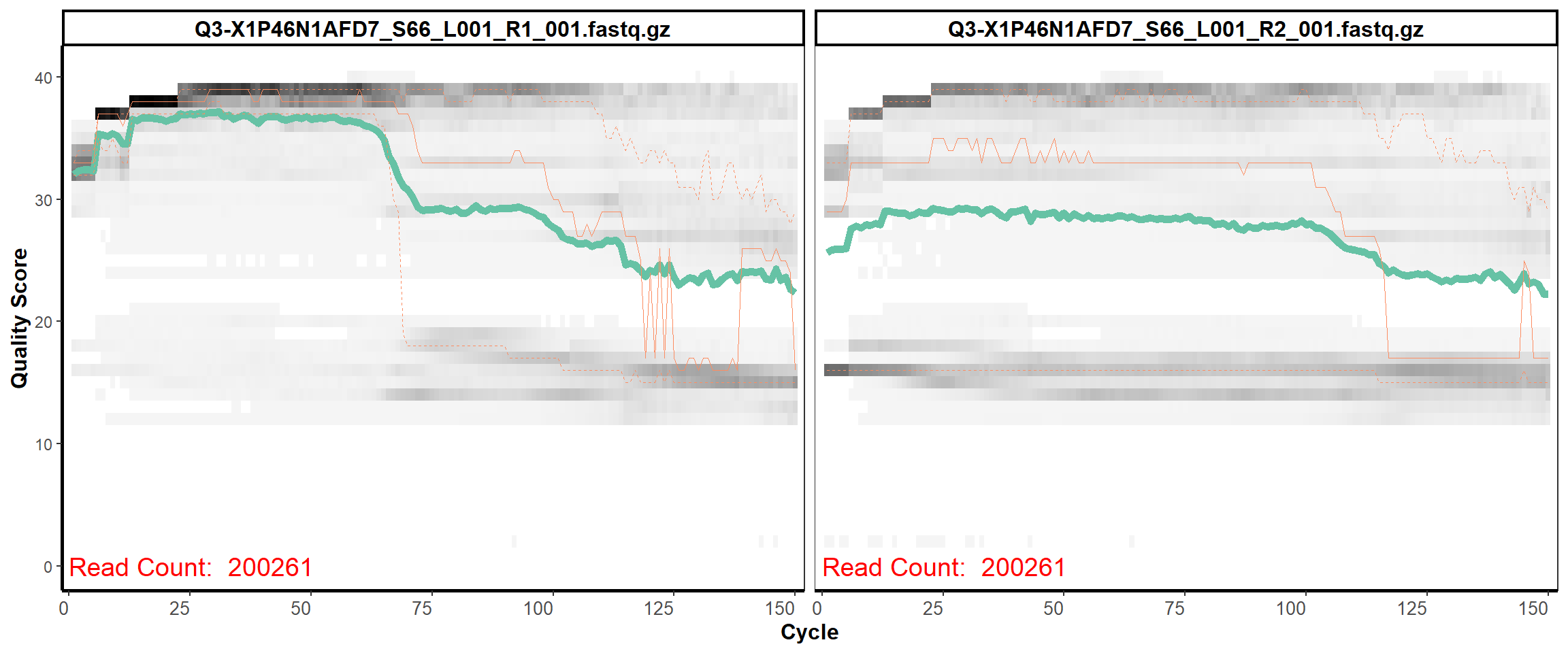
19
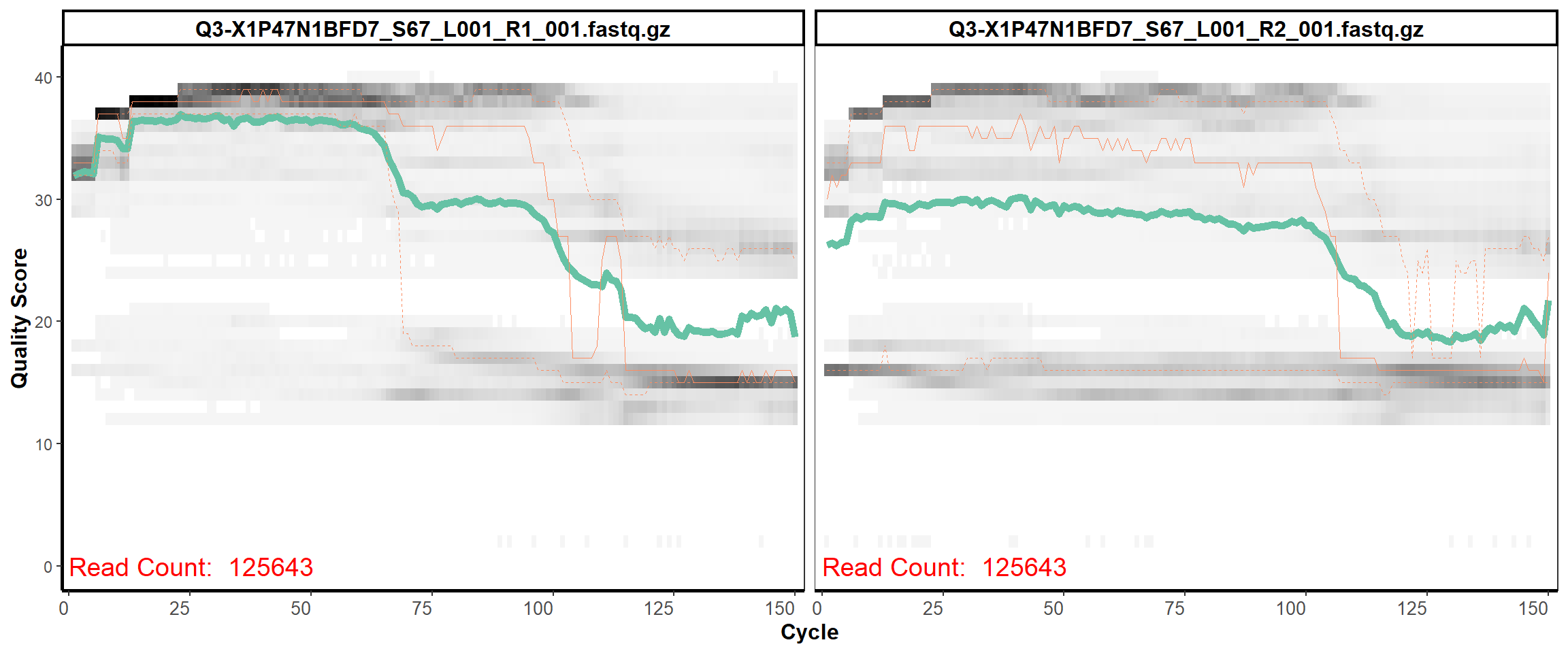
20

21
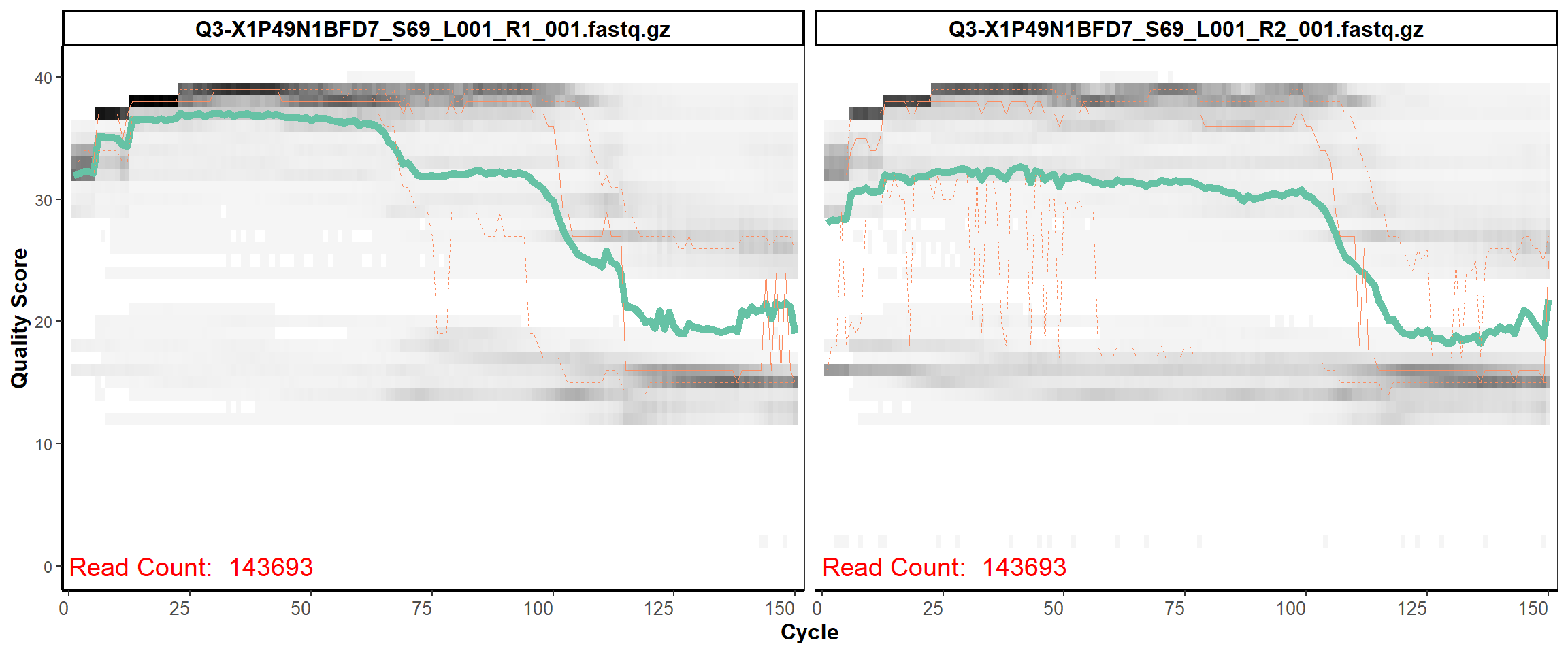
22
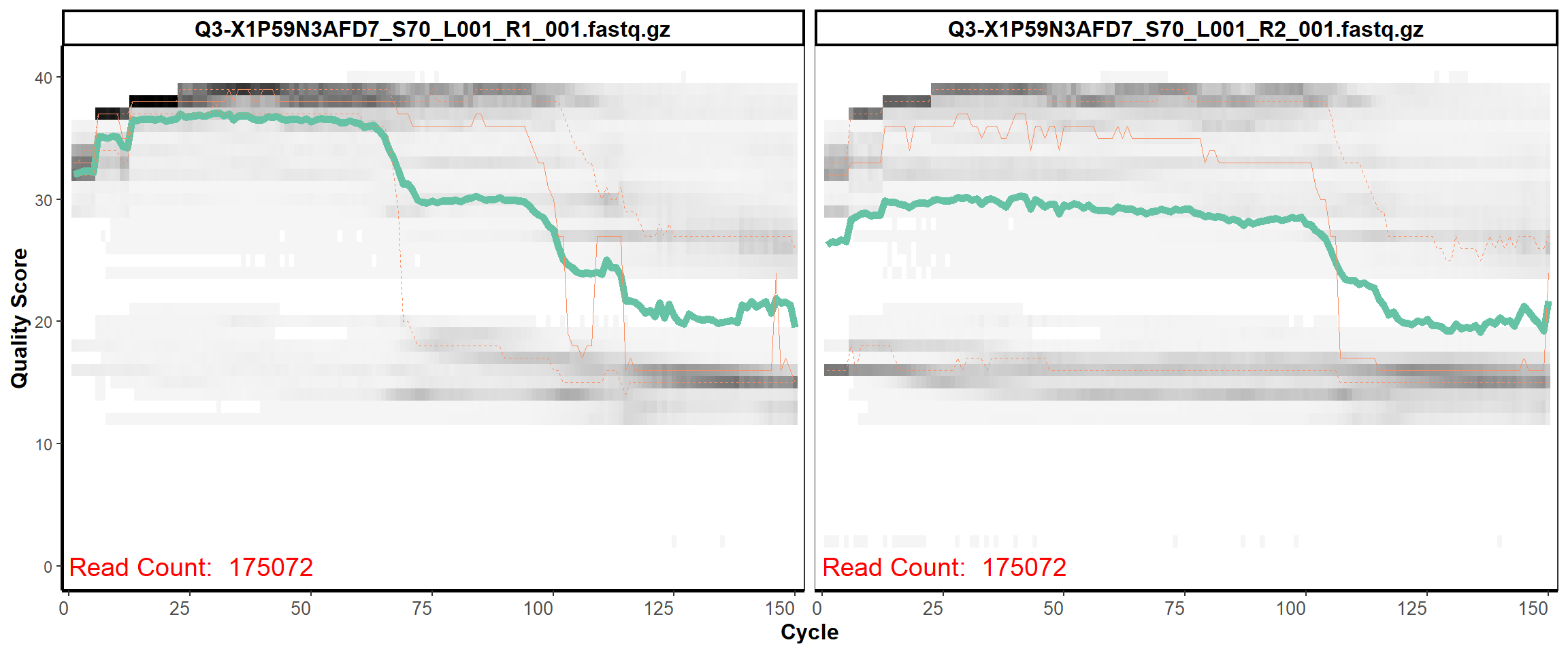
23

24
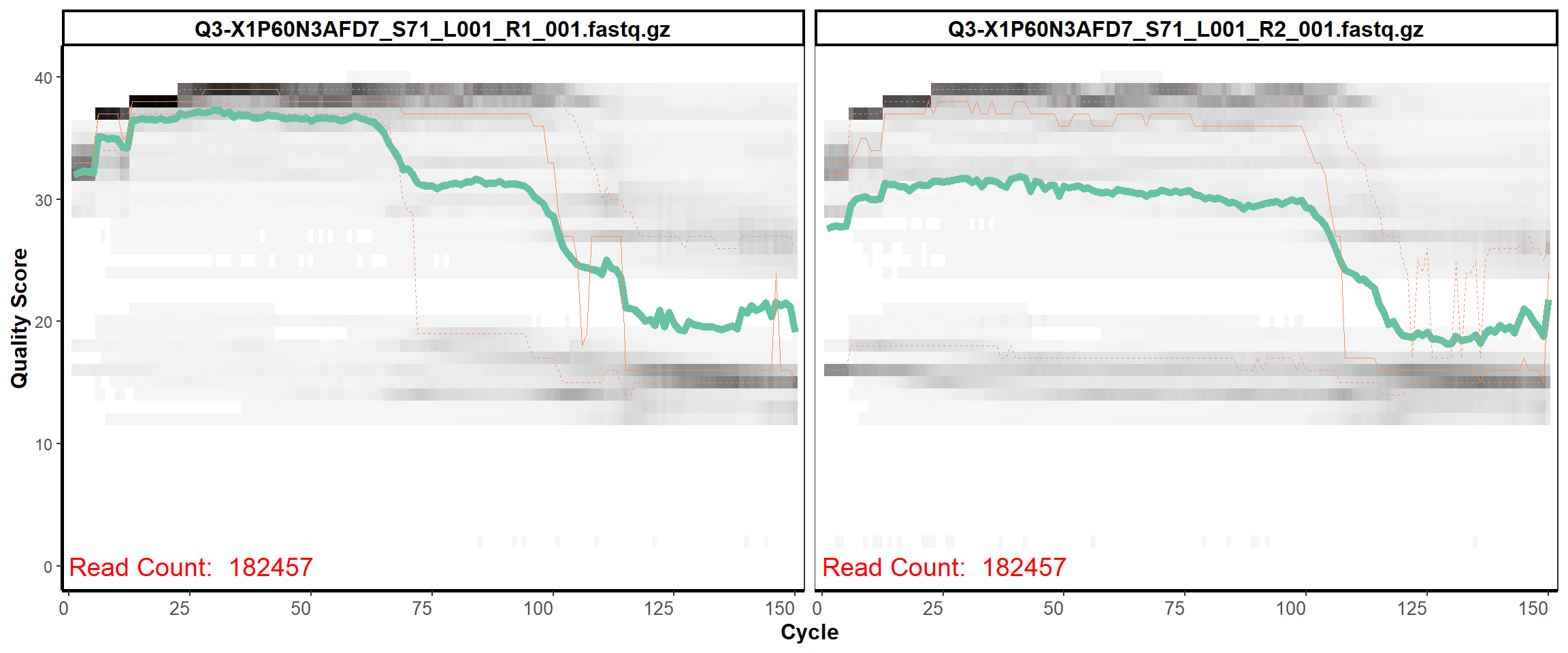
25
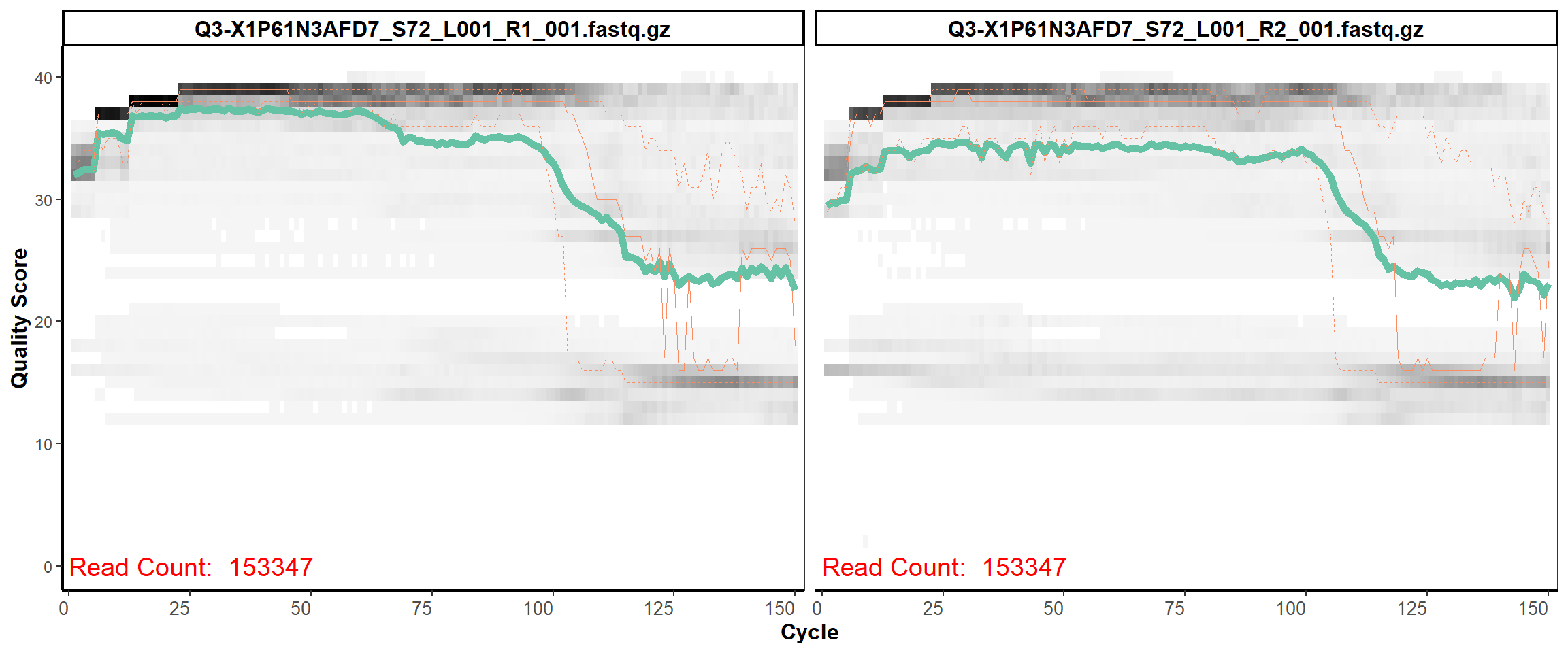
26
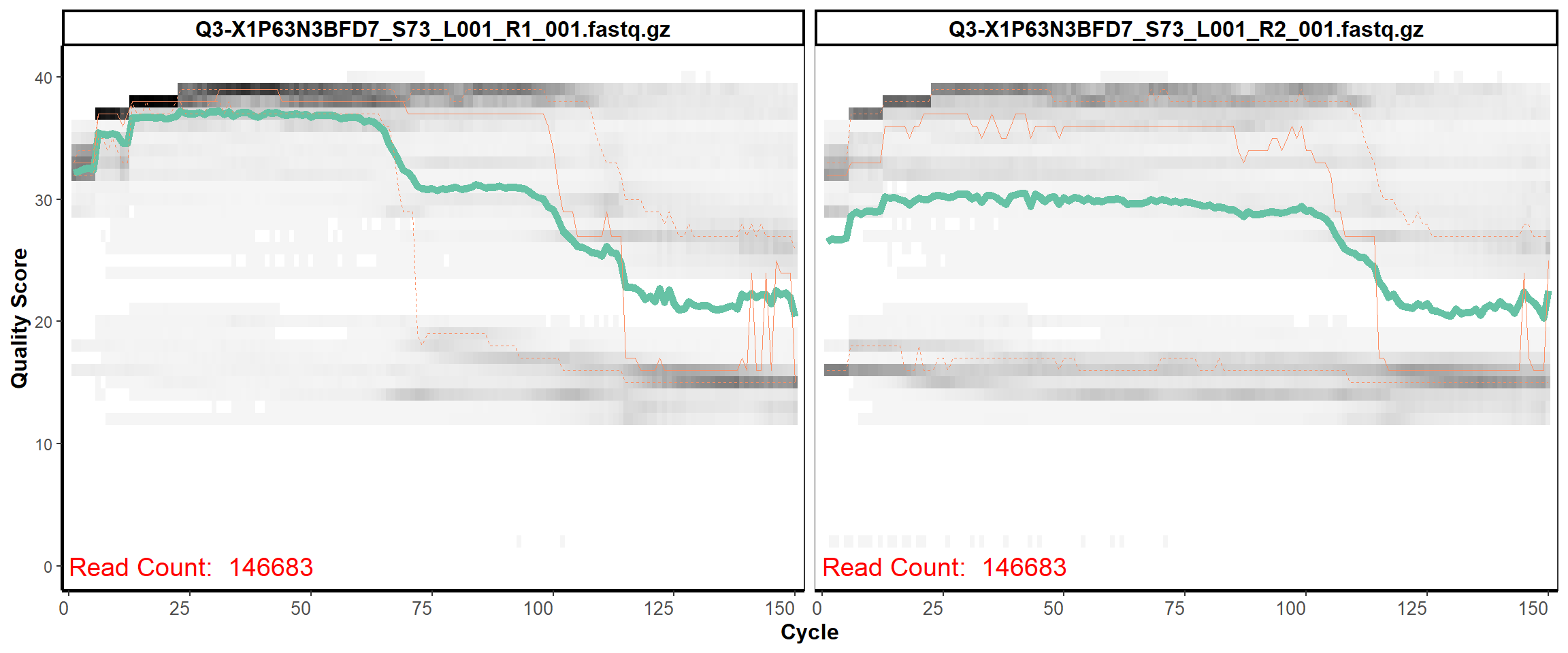
27
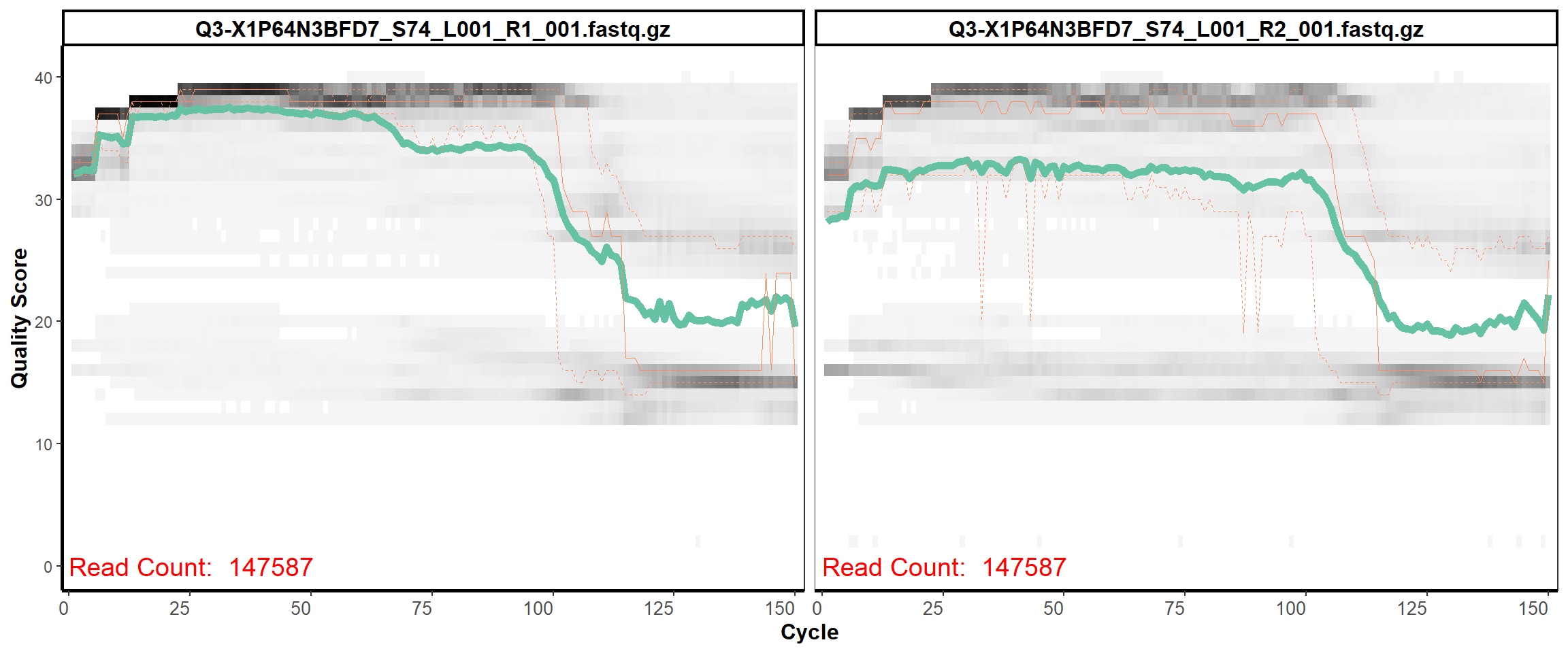
28

29
Sequence Results
Read Processing Pipeline
Workflow

*Demultiplexing done for analysis, though not necessarily required for practical implementation of DARTE-QM.
Software
Cutadapt - demultiplexing and primer-trimming
Cutadapt removes adapter sequences from high-throughput sequencing reads
PEAR - paired-end read assembler
PEAR: a fast and accurate Illumina Paired-End reAd mergeR
BLAST - basic local alignment search tool
Basic local alignment search tool
RDP-Classifier - 16S classifier
Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy
Primer Trimming and Demultiplexing
wd="../data/read_processing"
raw_files="${wd}/raw"
for raw_file in $(ls ${raw_files}/putvariablehere*_R1_*)
do \
sample="$(basename -- $raw_file)"
sample=$(echo "$sample" | cut -f 1 -d '.')
sample=$(echo "$sample" | cut -f 1 -d '_')
mkdir -p ${wd}/split/${sample}
cutadapt \
--pair-filter=both \
-e 0.10 \
-q 15 \
-g file:${wd}/../data/DARTE_forward_primers \
-G file:${wd}/../data/DARTE_reverse_primers \
-o ${wd}/split/${sample}/{name}.R1.1.fastq \
-p ${wd}/split/${sample}/{name}.R2.1.fastq \
$(ls ${wd}/raw/${sample}*_R2_*) \
$(ls ${wd}/raw/${sample}*_R1_*)
cutadapt \
--pair-filter=both \
-e 0.10 \
-q 15 \
-g file:${wd}/../data/DARTE_forward_primers \
-G file:${wd}/../data/DARTE_reverse_primers \
-o ${wd}/split/${sample}/{name}.R1.2.fastq \
-p ${wd}/split/${sample}/{name}.R2.2.fastq \
${wd}/split/${sample}/unknown.R2.1.fastq \
${wd}/split/${sample}/unknown.R1.1.fastq
donePair-end Read Merging
for primer in $(grep ">" ${wd}/../data/DARTE_forward_primers | cut -c2-)
do \
mkdir -p ${wd}/merge/${sample}
mkdir -p ${wd}/combined/${sample}
cat ${wd}/split/${sample}/${primer}*R1* > ${wd}/split/${sample}/${primer}.R1.fastq
cat ${wd}/split/${sample}/${primer}*R2* > ${wd}/split/${sample}/${primer}.R2.fastq
for file in $(ls ${wd}/split/${sample}/*.R1.fastq)
do \
file_name="$(basename -- $file)"
file_name=${file_name%.fastq}
file_name=$(echo $file_name| tr : _)
file_name=$(echo "$file_name" | cut -f 1 -d '.')
pear/bin/pear \
-j 8 \
-p 0.05 \
-v 10 \
-m 350 \
-f ${file} \
-r ${wd}/split/${sample}/${file_name}.R2.fastq \
-o ${wd}/merge/${sample}/${file_name}_merged
cat ${wd}/merge/${sample}/${file_name}_merged.assembled.fastq \
${wd}/merge/${sample}/${file_name}_merged.unassembled.forward.fastq \
${wd}/merge/${sample}/${file_name}_merged.unassembled.reverse.fastq \
> ${wd}/combined/${sample}/${file_name}_combined.fastq;
sed -n '1~4s/^@/>/p;2~4p' ${wd}/combined/${sample}/${file_name}_combined.fastq \
> ${wd}/combined/${sample}/${file_name}.fasta
done
doneBLAST
makeblastdb -in ../data/ARG_database.fa -dbtype nucl -out ../data/DARTE-QM_ARGs
for primer in $(grep ">" ${wd}/../data/DARTE_forward_primers | cut -c2-)
do
cat ${wd}/split/${sample}/${primer}*R1* > ${wd}/split/${sample}/${primer}.R1.fastq
cat ${wd}/split/${sample}/${primer}*R2* > ${wd}/split/${sample}/${primer}.R2.fastq
for file in $(ls ${wd}/split/${sample}/*.R1.fastq)
do \
file_name="$(basename -- $file)"
file_name=${file_name%.fastq}
file_name=$(echo $file_name| tr : _)
file_name=$(echo "$file_name" | cut -f 1 -d '.')
/mnt/home/smithsch/software/ncbi-blast-2.9.0+/bin/blastn \
-db ../data/DARTE-QM_ARGs \
-query ${wd}/combined/${sample}/${file_name}.fasta \
-out ${wd}/blast/${sample}/${file_name}.blast \
-perc_identity 90 \
-outfmt 6 \
-num_threads 8;
done
done
Schuyler Smith
Ph.D. Student - Bioinformatics and Computational Biology
Iowa State University. Ames, IA.